3-year-old boy with fever and cough

Case summary
A 3-year-old boy presented with a fever and cough that had been ongoing for 2 weeks. Physical examination revealed no abnormalities, and laboratory data were essentially normal. The patient had no postnatal respiratory symptoms.
Findings
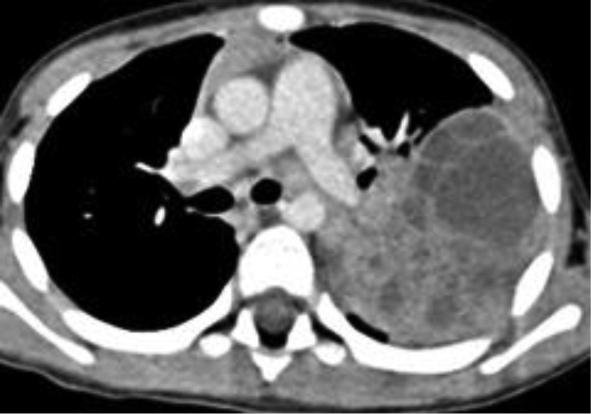
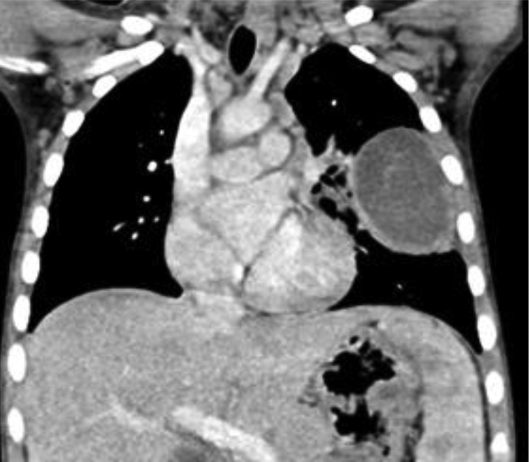
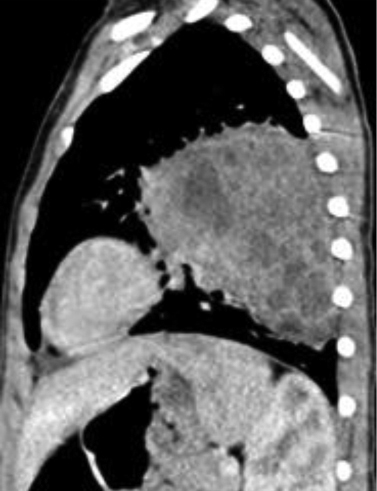
An initial chest radiography showed a cystic mass in the left upper lobe. A computed tomography (CT) scan was then performed with a Somatom Definition Flash scanner (Siemens, Erlangen, Germany). The CT scan revealed a large multicystic mass in the left upper lobe containing air-filled cysts measuring 2 cm or less in diameter and compression of adjacent lung parenchyma (Figures 1 and 2). Left upper lobectomy was performed.
Diagnosis
Congenital pulmonary airway malformation (CPAM, Stocker type 2), previously known as congenital cystic adenomatoid malformation (CCAM)
Discussion
CPAM is a hamartomatous lesion of the lung thought to result from abnormal distal airway branching.1 In Stocker et al, original classification,2 CPAM was known as CCAM and based on size of the cysts. It was divided into 3 major types: Type I (macrocystic, > 2 cm), Type II (multiple smaller cysts, < 2 cm), and Type III (solid with microscopic cysts). In the current expanded Stocker classification, these cystic lung lesions are now termed CPAMs.3 This classification includes the original Types 1, 2, and 3 and 2 additional Types (0 and 4). The lesions were renamed types 0-4 CPAM because cystic or adenomatoid features are not present in all lesions. CPAM usually communicates with the tracheobronchial tree, and has feeding arteries and draining veins from the pulmonary circulation. CPAM can arise in all lobes; except for with Types 0 and 3, it usually involves only one lobe. Types 0 and 3 can involve the whole lung.
Type 0 (< 3% of cases) arises from the trachea, has no cysts or cysts < 0.5 cm in size, involves all lobes, and is not compatible with life.3 Type 1 CPAM (60% to 70% of cases) arises from distal bronchial/proximal bronchiolar regions and is characterized by single or multiple cysts > 2 cm in size; Type 2 (15% to 20% of cases) arises from terminal bronchioles and has cysts < 2 cm in size; Type 3 (5% to 10% of cases) has an acinar origin and is predominantly solid with cysts < 0.5 cm; and Type 4 CPAM (10% to 15% of cases) has alveolar/distal acinar origins and contains large cysts > 2 cm in size, similar to Type 1 CPAM. Type 4 lesions can present with sudden respiratory distress due to tension pneumothorax and are strongly associated with malignancy, especially pleuropulmonary blastoma (1, 3, 4). Types 2 and 3 CPAM are commonly associated with other congenital anomalies, such as tracheoesophageal fistula, renal agenesis, intestinal atresia, and diaphragmatic hernia.
CPAM may be diagnosed on prenatal ultrasound. In the neonatal period, large lesions may cause respiratory distress. Smaller lesions may present months or years later as incidental findings on imaging studies or as recurrent pulmonary infection.
Chest radiography is the initial imaging examination in the diagnosis of CPAM. CT is used for confirmation of the diagnosis, characterization of the lesion, and determination of extent for surgical planning. On CT, Type 1 CPAM appears as a multicystic lesion with one or a few large cysts > 2 cm; the cysts may be air-filled or have air-fluid levels. Type 2 CPAM contains numerous small cysts < 2 cm in size. Type 3 CPAM appears as a large, solid, homogeneous mass. Type 4 CPAM has an appearance similar to Type 1, but there may an associated pneumothorax. In all types, associated findings include mediastinal shift to the contralateral side and hypoplasia of the ipsilateral lung.5,6
The differential diagnosis of CPAM includes congenital diaphragmatic hernia, bronchogenic cyst, lobar emphysema, bronchogenic cyst, and localized pulmonary interstitial emphysema that may arise in patients receiving mechanical ventilation, and pneumatocele resulting from bacterial pneumonia, especially due to staphylococcal infection. Of note, CPAM and sequestration can coexist; presence of systemic arterial supply should suggest diagnosis of a combined lesion.
Bronchioloalveolar carcinoma and rhabdomyosarcoma, in addition to pleuropulmonary blastoma, have been reported in association with CPAM/CCAM.7 Because there is an association with malignancy and risk of recurrent infections, the recommendation is usually resection, even in asymptomatic patients.
References
- Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17-37.
- Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung: Classification and morphologic spectrum. Hum Pathol. 1977;8:155-171.
- Stocker JT. Congenital pulmonary airway malformation: A new name and an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology. 2002;41(suppl 2):424-431.
- Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol. 2009;28:155-184.
- Griffin N, Devaraj A, Goldstraw P, et al. CT and histopathological correlation of congenital cystic pulmonary lesions: A common pathogenesis? Clin Radiol. 2008;63: 995-1005.
- Shimohira M, Hara M, Kitase M, et al. Congenital pulmonary airway malformation: CT-pathologic correlation. J Thorac Imaging. 2007;22:149-153.
- Lakhoo K. Management of congenital cystic adenomatous malformations of the lung. Arch Dis Child Fetal Neonatal Ed. 2009; 94:F73–F76.