Pancreatoblastoma
By Cornejo P, Jorgensen S, Towbin R
CASE SUMMARY
A 5-year-old boy with no significant past medical history presented to the emergency department with abdominal distention and pain. A laboratory evaluation showed elevated lactate dehydrogenase (LDH, 447 U/L] and alpha-fetoprotein (AFP, 6690 ng/mL].
IMAGING FINDINGS
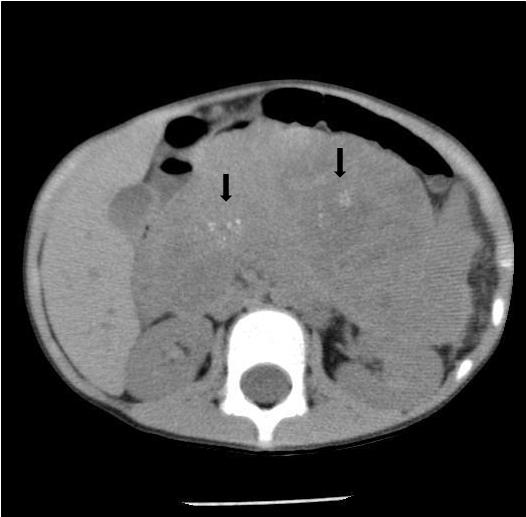
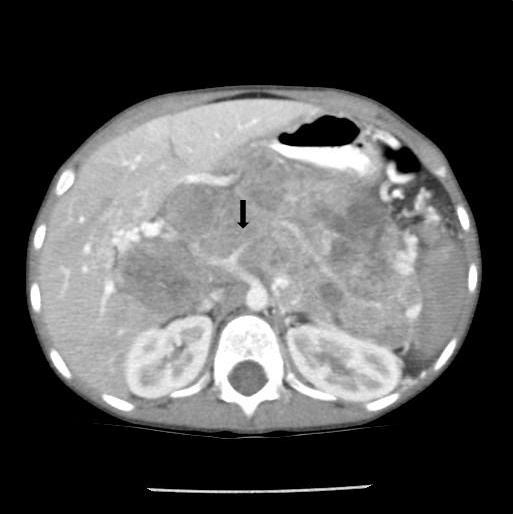
Noncontrast abdominal CT demonstrated a 13 × 10 × 9 cm midline retroperitoneal mass extending into the mid abdomen. The mass was heterogeneous with multiple small calcifications (Figure 1). There was mass effect on the liver, spleen, left kidney, and adjacent bowel. No normal pancreas tissue was identified. Follow-up contrast-enhanced CT showed mild patchy enhancement. The mass encased the celiac axis (Figure 2). The superior mesenteric artery, aorta, and IVC were not involved by tumor. Numerous gastric varices, porta hepatis and splenic hilum collaterals were noted. No additional abnormalities were present. A whole-body nuclear medicine bone scan (not shown) demonstrated no osseous metastatic disease. An open biopsy subsequently determined that the tumor was unresectable.
Histologic analysis demonstrated sheets, strands, and nests of two types of cells: gland-forming acini-like and squamous cells in an organoid arrangement. There was evidence of vascular invasion. The tumor was positive for AFP and CD10. Tumor cells demonstrated synaptophysin, AE1/AE3 (pancytokeratin), beta-catenin, and alpha-1-antitrypsin (α-1-antitrypsin) staining.
The patient started chemotherapy with vincristine, doxorubicin, and cyclophosphamide (mesna therapy). G-CSF therapy was also started.
DIAGNOSIS
Pancreatoblastoma
DISCUSSION
Pancreatoblastoma is the most common primary pancreatic neoplasm in early childhood, originating from acinar (exocrine) cells.1,2 The tumor usually presents in the first decade of life with a mean age of 5 years, but rare cases have been reported in adults.3,4 The tumor occurs twice as often in males, with more than half of cases reported in individuals of East Asian descent.2 Beckwith-Wiedemann syndrome (BWS) and familial adenomatous polyposis have been associated with congenital pancreatoblastomas.1,4 The congenital forms in BWS are often cystic in nature.
The clinical presentation and symptoms are usually nonspecific. Abdominal distention, pain, and failure to thrive are common. In some cases, tumors secrete hormones;thus, Cushing syndrome or inappropriate antidiuretic hormone may be the initial presentation.4 AFP is the most common elevated serological marker; however, LDH, α-1-antitrypsin, glucose-6-phosphatase, and CA 19-9 may also be abnormal.4
Pancreatoblastomas can involve the head, body or tail of the pancreas, although body/tail or the whole organ are most commonly involved.2 Due to the central location of the pancreas, direct extension of the tumor to the liver, spleen, left kidney, left adrenal gland, and omentum can occur.2 Vascular encasement is common, particularly the mesenteric vessels, IVC, and portal vein.2 The aorta is rarely involved. This helps in differentiating pancreatoblastomas from other intra-abdominal masses such as neuroblastoma.
Other entities in the differential diagnosis include Wilm’s tumor, hepatoblastoma, solid and cystic papillary tumor of the pancreas, non-Hodgkin lymphoma, and a Kaposiform hemangioendothelioma.1,2 The age and gender of the child should help to narrow down the differential diagnosis. Large tumors, as in our case, make identification of the site of origin difficult.
Pancreatoblastomas are slow-growing masses and often remain occult until they are large. As a result, approximately 35% of patients have liver and lymph-node metastases at presentation.1 The liver is the most common site of metastatic disease.3 Other less common locations of metastases include the lungs, brain, spleen, kidney, adrenal gland, omentum, and bone.1,2
Ultrasound, CT and MRI are utilized for diagnosis and surveillance. All three modalities are well suited for tumor evaluation. On ultrasound, the mass has well-defined margins with solid and cystic components.1,2 The tumor is vascular; therefore, Doppler interrogation is an important component of the exam.
CT will demonstrate a multilobulated mass that is hypodense to the liver.1,2 The tumor usually has a heterogeneous appearance as a result of cyst formation or central necrosis. Scattered punctate or clustered calcifications may be found.1 Mild enhancement of vascular structures and septations within the mass will be noted on postcontrast imaging. When present, liver metastases are hypodense.1 Dilation of the intra- and extrahepatic biliary ducts is rarely seen since the tumor has a soft consistency with no significant ductal compression.1
On MRI, the tumor is usually heterogeneous with well-defined margins. On T1-weighted images the tumor has low to intermediate signal while on T2-weighted images, the tumor has heterogeneous high signal compared to the liver. After contrast, mild patchy enhancement is seen.1,2
The goal of treatment is complete surgical resection. Long-term survival is possible with complete resection, although, pancreatoblastomas have a high recurrence rate.4 When primary resection is not possible; neo-adjuvant chemotherapy is the preferred treatment. Indications for neo-adjuvant chemotherapy include large tumors that involve adjacent major vessels, solid organs, and metastatic disease.4 Chemotherapy alone can be useful for unresectable, metastatic or recurrent masses. Radiotherapy is used only in selected, inoperable cases.
Long-term follow-up is necessary to identify and treat recurrence and metastasis. High-risk patients with BWS and familial adenomatous polyposis should be screened regularly with ultrasound and cross-sectional imaging for suspicious findings.4
CONCLUSION
Pancreatoblastomas are the most common pancreatic neoplasms in early childhood. The tumor symptoms are often nonspecific. Ultrasound, CT and MRI are complementary in tumor diagnosis and follow-up treatment. Patients with Beckwith-Wiedemann syndrome and familial adenomatous polyposis have an increased risk for congenital pancreatoblastomas and should be screened regularly.
REFERENCES
- Chung EM, Travis MD, Conran RM. Pancreatic tumors in children: Radiologic-pathologic correlation. RadioGraphics. 2006; 26:1211-1238.
- Roebuck DJ, Yuen MK, Wong YC, et al. Imaging features of pancreatoblastoma. Pediatr Radiol. 2001;31:501-506.
- Montemarano H, Lonergan GJ, Bulas DI, et al. Pancreatoblastoma: Imaging findings in 10 patients and review of literature. Radiology. 2000; 214:476-482.
- Glick RD, Pashankar, FD, Pappo A, et al. Management of pancreatoblastoma in children and young adults. J Pediatr Hematol Onco. 2012; 34 Suppl 2:S47-50.