Radiological Case: Imaging findings on heparin-induced thrombocytopenia with thrombosis

CASE SUMMARY
A 53-year-old woman diagnosed with ovarian cancer was transferred to the ICU requiring mechanical ventilation and pressure support. In the 2 days prior to admission she experienced nausea and vomiting that started soon after the placement of a chest wall port catheter with a baseline platelet count of 338,000 /mm3 (Figure 1). After a complicated clinical course which required heparin drip for myocardial infarction, the patient experienced upper GI bleeding and a cold left foot with absent pulse in the dorsalis pedis artery. On day 9 from initial exposure to heparin, clinical suspicion of heparin-induced thrombocytopenia with thrombosis (HITT) led to evaluation with HIT antibodies which were confirmed days later by an enzyme-linked immunosorbent assay (ELISA).
Cross-sectional imaging (Figures 2 and 3) performed on days 10 and 11 demonstrated evidence of wide spread thrombosis. On day 11, argatroban was initiated in lieu of heparin, however, the patient died on day 14 from initial exposure to heparin.
IMAGING FINDINGS
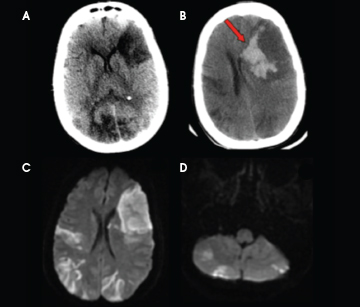
An unenhanced head CT obtained on day 10 from initial exposure to heparin for persistent altered mental status demonstrated multifocal, wedge-shaped hypodensities involving both grey and white matter in keeping with ischemic stroke which was later corroborated with MRI (Figure 2). DWI MR images showed restricted diffusion in the corresponding hypodense regions on CT as well as additional stroke burden involving the posterior circulation (Figure 2). Hemorrhagic conversion was demonstrated in Figure 2B obtained as an unenhanced CT 2 days after the initial head CT.
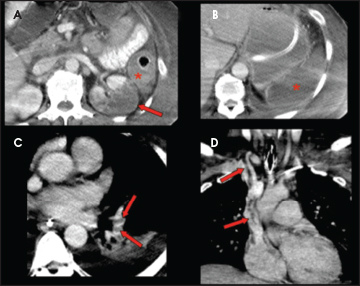
On day 11 from initial exposure to heparin, a contrast-enhanced CT of the chest, abdomen and pelvis was performed (Figure 3) and demonstrated thrombosis within the right internal jugular vein with extension into the superior vena cava and right atrium. Additionally, bilateral pulmonary emboli and extensive solid organ infarction affecting the spleen and kidneys in addition to colonic ischemia were also demonstrated (Figure 3).
DIAGNOSIS
Heparin-induced thrombocytopenia with thrombosis (HITT)
Differential diagnosis: Cardiogenic emboli, venous thromboembolism and disseminated intravascular coagulation (DIC)
DISCUSSION
Heparin-induced thrombocytopenia (HIT) is a relatively common complication affecting approximately 5%-30% of patients exposed to heparin.1 Despite a decrease in platelet count, 20%-60% of patients with HIT develop thrombotic complications,2,3 also known as heparin-induced thrombocytopenia with thrombosis (HITT) or white clot syndrome. HIT has been classically defined as an absolute reduction in platelet count below 100,000/mm3 or a relative decrease of 50% or more from baseline.1,2 Since its first description in the late 1940’s three types of HIT have been described, two of which present with asymptomatic thrombocytopenia.1 The third type of HIT is associated with clinically significant arterial and/or venous thrombosis (HITT) with consequent high morbidity and mortality.3,4
HIT is an immune mediated reaction to heparin and, even though it is considered as unpredictable, some risk factors have been described. These include exposure to heparin for more than 5 days, being on hemodialysis, and having a diagnosis of autoimmune disease, gout or heart failure.5 Nonetheless, thrombotic complications only develop in a subgroup of patients with HIT. In a study of 408 patients with HIT, 40% developed pulmonary embolism, and it was shown that the most important risk factors for thrombosis were orthopedic/trauma surgery and the magnitude of platelet count decrease.6
The clinical diagnosis of HITT is suspected in any patient with thrombocytopenia that is time-concordant with heparin exposure and after the exclusion of other causes of thrombocytopenia such as infection/sepsis, drug-induced, disseminated intravascular coagulation (DIC) and immune thrombocytopenic purpura (ITP), platelet recovery after discontinuation of heparin, and the development of limb ischemia or end-organ dysfunction from arterial and/or venous thrombosis.1,7 Once HITT is suspected clinically, immediate action should be taken by discontinuing heparin and switching to a direct thrombin inhibitor (DTI). In the mean time, a laboratory work-up can be initiated with the use of ELISA, which detects circulating antibodies that promote the formation of IgG/platelet factor 4/heparin complex which enhances platelet activation and aggregation that, in turn, lead to thrombocytopenia and, in a selected subgroup, thrombosis.1, 2 The platelet aggregation test and the carbon-14-serotonin release assay are other ways of demonstrating in vitro heparin-dependent platelet antibodies that have been described.1
The imaging findings of HITT rely on the identification of a vascular filling defect that is in keeping with intraluminal clot and/or the imaging sequelae of end-organ thrombosis. For example, in the brain, this is manifested as loss of the grey-white matter differentiation or restricted diffusion in a typical vascular distribution; in the kidney, the classic wedge-shaped areas of decreased attenuation or enhancement can be seen. While these findings are specific for infarction or ischemic change the etiologic factor is often not derived from the imaging study as they are similar to those caused by venous thromboembolic disease, cardiogenic embolism, or any prothrombotic state such as malignancy and DIC. However, in the setting of both arterial and venous thrombosis the consideration of HITT should be strongly entertained and, even in the absence of venous thrombosis, the presence of multifocal arterial thrombosis should also raise this possibility. Some of the complicating features of this life-threatening condition that have been described in the radiology literature include: cerebral stroke, myocardial infarction, aortic and mesenteric thrombosis, pulmonary emboli, extremity thrombosis, solid organ infarction, and deep venous thrombosis.3,4,8,9 While most of the complications in HITT are a result of thrombosis, hemorrhagic sequelae, as seen on this case, have also been described.3,4
The goal of management in patients with HIT is to reduce the thrombotic risk by reducing platelet activation and thrombin generation.2 This can be accomplished by prompt discontinuation of all sources of heparin, including heparin lock flush IV solutions, and initiating treatment with a DTI such as argatroban.2,10 Despite these measures the mortality rate in HITT remains high (8%-36%).1,2
CONCLUSION
The clinical diagnosis of HITT should be suspected in anyone with thrombocytopenia that is time-concordant with exposure to heparin and has evidence of end-organ dysfunction. Early diagnosis is imperative since this antibody-mediated drug reaction is associated with high morbidity and mortality and because one of the cornerstones of treatment involves immediate discontinuation of the offending drug. Thus, a high clinical suspicion is essential for establishing a prompt diagnosis.
Even though the imaging findings of HITT are nonspecific, given the widespread use of noninvasive imaging techniques to evaluate medically complex patients, the radiologist should consider this infrequent entity in the setting of widespread end-organ infarction, especially when both arterial and venous thrombosis are present. By recognizing these findings the radiologist may be the first in suspecting HITT and should recommend correlation with platelet count and, if clinically warranted, HIT antibodies.
Disclaimer: The opinions and assertions contained herein are the private views of the authors and are not to be construed as official nor as representing the views of the Departments of the Army, Navy, Air Force, or Defense.
REFERENCES
- Murphy KD, Galla DH, Vaughn CJ, et al. Heparin-Induced Thrombocytopenia and Thrombosis Syndrome. Radiographics. 1998;18:111-120.
- Arepally GM, Ortel TL. Heparin-Induced Thrombocytopenia. N Engl J Med. 2006;355:809-817.
- Silver D, Kapsch DN, Tsoi EKM. Heparin-Induced Thrombocytopenia, Thrombosis, and Hemorrhage. Ann Surg. 1983;198:301-305.
- Rhodes GR, Dixon RH, Silver D. Heparin Induced Thrombocytopenia: Eight Cases with Thrombotic-Hemorrhagic Complications. Ann Surg. 1977;186: 752-758.
- Kato S, Takahashi K, Ayabe K, et al. Heparin-induced thrombocytopenia: analysis of risk factors in medical inpatients. Br J Haematol. 2011;154: 373-377.
- Greinacher A, Farner B, Kroll H, et al. Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb Haemost. 2005;94:132-135.
- Sekhon SS, Roy V. Thrombocytopenia in Adults: A Practical Approach to Evaluation and Management. Southern Medical Journal. 2006;99:491-497.
- Lindsey SM, Maddison FE, Towne JB. Heparin-Induced Thromboembolism: Angiographic Features. Radiology. 1979;131:771-774.
- Lammering JC, Wang DS, Shin LK. Heparin-induced thrombocytopenia (HIT) causing portosplenic, superior mesenteric, and splenic vein thrombosis resulting in splenic rupture and pulmonary emboli formation. Clin Imaging. 2012;36(6):865-868.
- Linkins LA, Dans AL, Moores LK, et al. Treatment and Prevention of Heparin-Induced Thrombocytopenia: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clincal Practice Guidelines. Chest. 2012;141:495-530.