Retroperitoneal Paraganglioma
By Gubler JT, Towbin R, Schaefer CM, Towbin AJ
Case Summary
A heathy teenager presented with left flank pain and dizziness after a collision with another player during a sporting event. Physical examination revealed diffuse left flank tenderness without ecchymosis. Vital signs were stable, with a blood pressure of 113/65 and heart rate of 66 bpm. Hemoglobin and hematocrit were normal.
Imaging Findings
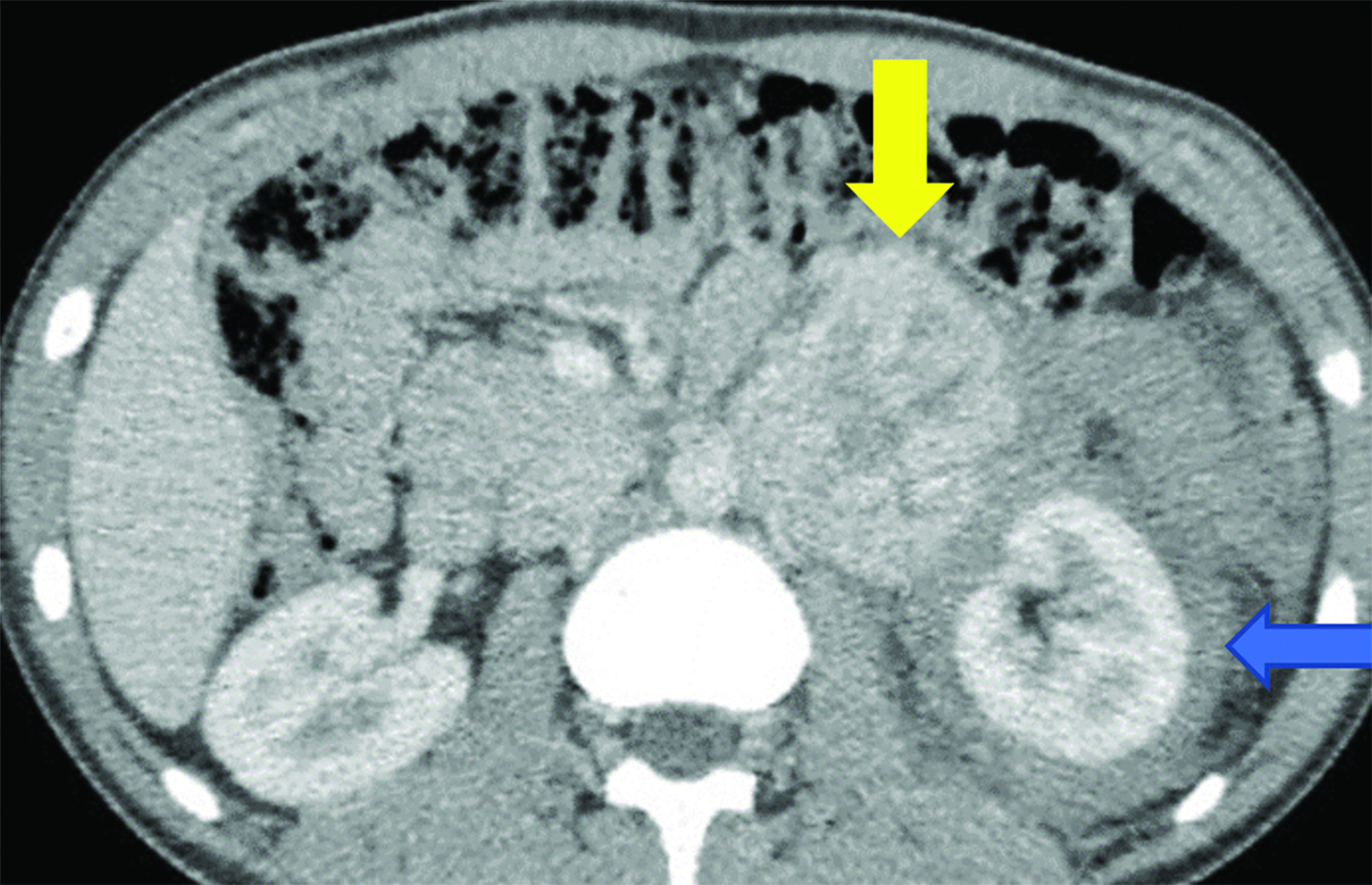
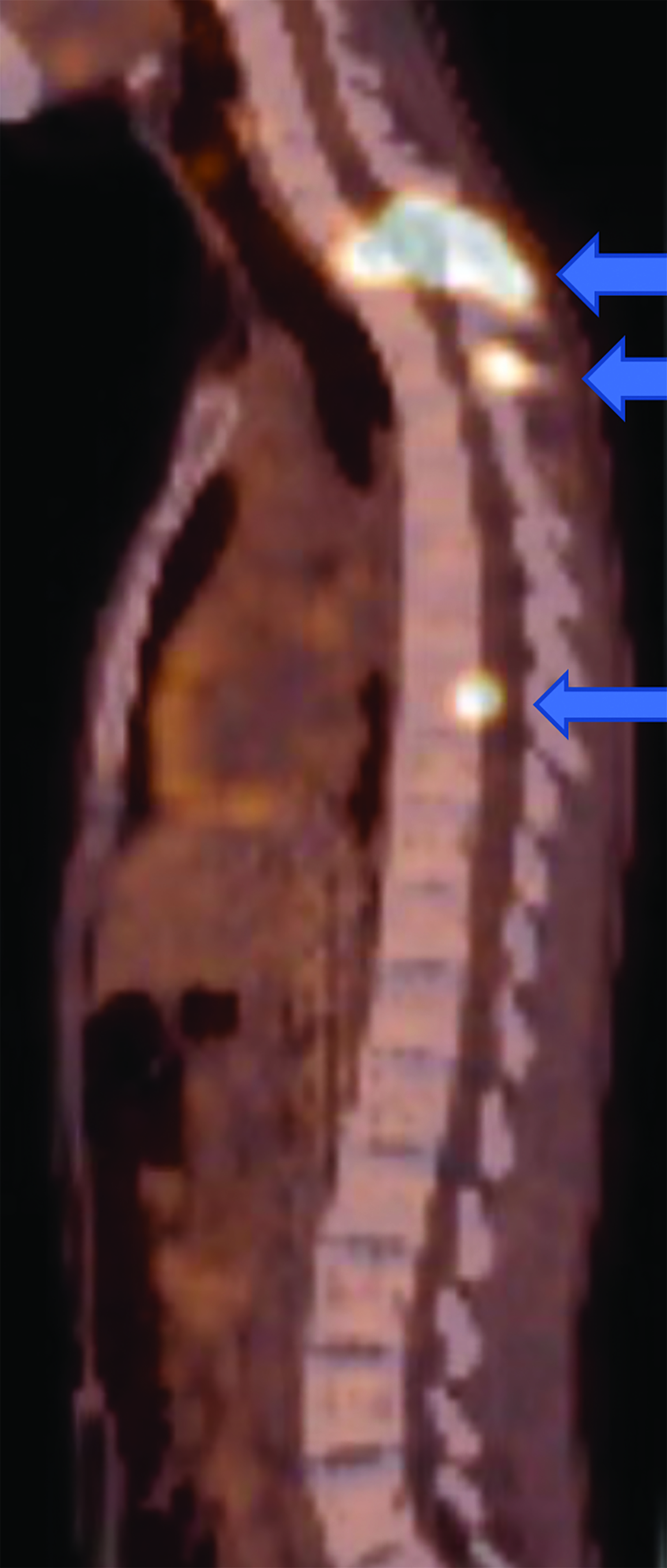
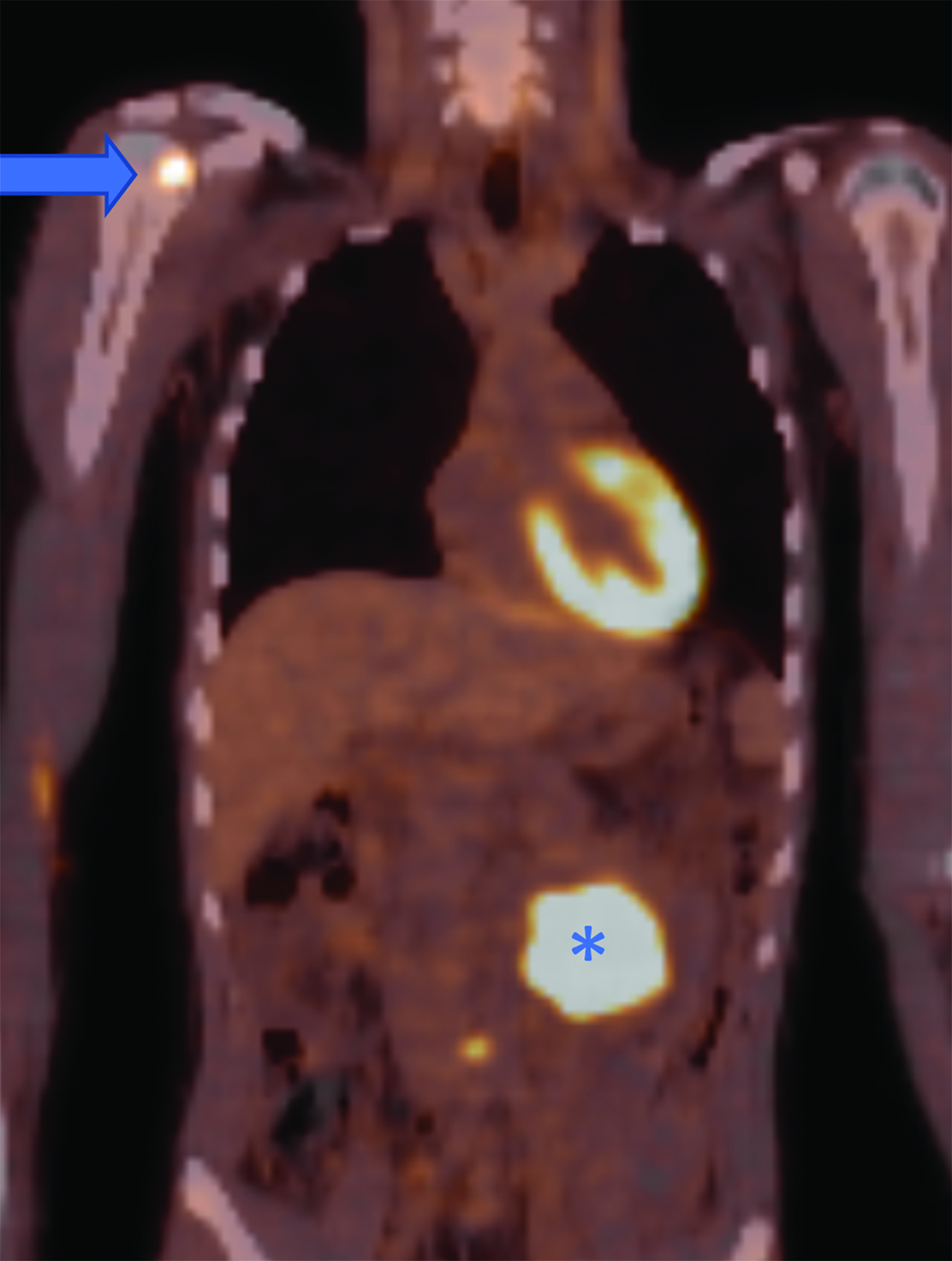
CT of the abdomen and pelvis demonstrated an enhancing, 7.5 cm, left retroperitoneal mass with central hypodensity, and a left perinephric and retroperitoneal hematoma (Figure 1). PET/CT showed uptake in the retroperitoneal mass with a maximum SUV of 15.6 and fluorodeoxyglucose-avid metastases to the skull, thoracic spine, and right humerus (Figure 2). The tumor type was confirmed preoperatively with an image-guided percutaneous biopsy.
Diagnosis
Retroperitoneal paraganglioma (PGL).
The differential diagnoses include lymphoma, metastasis (such as from a testicular tumor), and sarcoma.
Discussion
Paragangliomas and pheochromocytomas are rare primary retroperitoneal tumors composed of neuroendocrine cells, or chromaffin cells, that arise from neural crest cells known as pheochromoblasts. PGL arises in extra-adrenal sites, while pheochromocytoma arises in the adrenal medulla. The first documented PGL was described by Alezais and Peyron in 1908.1
It has been estimated that PGLs and pheochromocytomas occur in roughly 2-8 per 1 million people each year.2 PGLs can occur anywhere paraganglia are located, commonly in parasympathetic tissue in the head, neck, and thorax, or in sympathetic tissue in the abdomen and pelvis. PGLs most often occur sporadically, but approximately one-third are associated with genetic factors including von Hippel-Lindau Syndrome, multiple endocrine neoplasia types 2A and 2B, neurofibromatosis type 1, and succinate dehydrogenase complex mutations. Genetic testing can therefore be useful in diagnosing and managing suspected PGLs.
Clinical features are often related to the effects of the products of the tumor; namely excess catecholamines. Symptoms will fluctuate based on the levels of epinephrine, norepinephrine, and dopamine, and are therefore relapsing and remitting in nature. PGLs can present with symptoms classically described as the 5 P’s: increased blood pressure, head pain (headache), perspiration, palpitations, and pallor.
Other less-specific features include abdominal pain, anxiety, weight loss, and hyperglycemia. However, 20-30% or more of patients with PGLs have only minor signs or are asymptomatic, resulting in underdiagnosis.1
Diagnosing PGL relies on laboratory tests and imaging. Initially, plasma free-metanephrine tests such as homovanillic acid or vanillylmandelic acid can be utilized. A more specific and definitive biochemical approach is to test for metanephrines and catecholamines in a 24-hour urine specimen, with urinary vanillylmandelic acid having a 95% specificity.3 After a diagnosis of PGL is established, imaging is used to localize the tumor.
Several imaging modalities can be used to localize PGL. These include CT, MRI, single-photon emission computed tomography (SPECT) using 123I-metaiodobenzylguanidine (123I-MIBG) or 111In-DTPA-pentetreotide, and positron emission tomography using [6-[18F]-fluorodopamine (18F-FDA), 6-[18F]-fluoro-L-3,4-dihydroxyphenylalanine, and 2-[18F]-fluoro-2-deoxy-D-glucose (18F-FDG). Of these modalities, CT or MRI is considered first line. These approaches provide high sensitivity and typically find the tumor. Computed tomography of the neck, chest, abdomen, and pelvis is often used to identify the primary tumor and to assess for metastasis.
Coupling anatomical imaging with functional imaging specific to PGLs is necessary to provide specificity. Nuclear medicine scanning is especially useful owing to the unique characteristics of PGLs in taking up specific radiotracers. The use of 18F-FDG PET or 18F-FDA PET for suspected metastasis and 123I-MIBG SPECT for established metastasis are most useful; however, other markers should be considered based on the entire clinical picture.4
Approximately 25% of PGLs are malignant.5 Malignancy is determined by the tumor’s behavior; ie, metastasis to non-chromaffin tissue, and not necessarily its histology. Histology of PGLs, though, typically shows circumscribed nests of polyhedral to fusiform neoplastic cells, known as “zellballen,” that contain granular, amphophilic or basophilic cytoplasm and vesicular nuclei.6
Prediction of malignancy is often difficult and involves several risk factors and a scoring system. Risk factors include succinate dehydrogenase complex gene mutations, primary tumor size, younger age, and elevated 3-Methoxytyramine levels. The most extensively used scoring system is Grading of Adrenal Pheochromocytoma and Paraganglioma which grades PGLs as low, intermediate, or high risk for metastasis. Malignant PGLs typically spread to regional cervical lymph nodes or distant locations such as the bone, liver, and lungs, depending on their primary location above or below the neck, respectively.2
Treatment options and prognosis depend on whether the tumor is benign or malignant. Surgical removal is definitive for benign lesions; however, metastatic tumors are more complicated and often involve control of symptoms and tumor growth. Percutaneous image-guided biopsy may be valuable in confirming the diagnosis. However, it is essential to block the catecholamine effect before biopsy.
Symptomatic control for both benign and metastatic PGLs is obtained first by alpha and, when indicated, beta-adrenergic blockade. Phenoxybenzamine is a long-acting, nonselective, noncompetitive alpha-adrenergic blocker and is the most commonly used agent, followed by doxazosin, a selective alpha-1-adrenergic blocker. Slowly progressive tumors are sometimes monitored in cases of asymptomatic metastatic PGLs, but they require intensive lab and radiological following.7
Surgical resection is often not curative in metastatic PGLs; however, it is useful in controlling symptoms. Total surgical resection of the primary tumor, regional lymph nodes, and distant metastases is the preferred method. Radiofrequency ablation and direct intra-tumoral injection of absolute alcohol into unresectable tumors is also an option. Chemotherapy, molecular targeted therapy, and radionuclide therapy are often used in combination with multiple forms of treatment for adequate systemic therapy.
Prognosis in cases of metastases to the liver and lungs is generally poor, with shorter survival compared to metastases to the bone. Metastases to the bone develop in roughly 70% of these cases. These are mainly lytic lesions and require combination therapy with medications such as bisphosphonates or RANKL inhibitors, irradiation, and radiofrequency ablation.7
Long-term follow-up is required with any PGL or pheochromocytoma. Biochemical testing is performed to confirm complete tumor resection and further biochemical assays are used to monitor recurrence. Recurrence rates ranging from 14% to 30% have been documented, and malignancy is seen in half of recurrences.2,5 Therefore, life-long follow-up of these patients is appropriate.