Congenital adrenal hyperplasia secondary to 3-beta hydroxysteroid dehydrogenase deficiency
Images
CASE SUMMARY
A 35-year-old Hispanic man presented to the emergency department with a two-day history of abdominal pain, non-bilious vomiting and acute anorexia.
The patient was previously diagnosed with hypospadias and congenital adrenal hyperplasia (CAH) but has been without treatment for 3 years, due to lack of insurance. Surgical, family and social history were non-contributory.
Review of systems was negative, including no weight loss and no fever. Physical exam was within normal limits.
IMAGING FINDINGS
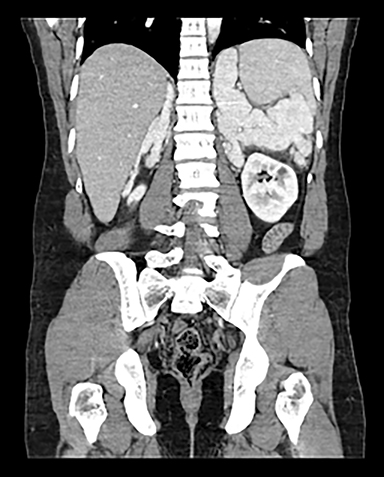
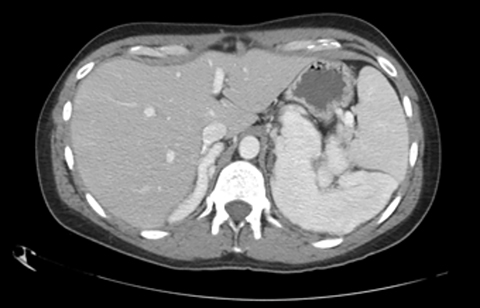
Contrast-enhanced computed tomography (CECT) of the abdomen and pelvis demonstrates lobulated homogenously enhancing masses measuring up to 10 cm in the bilateral adrenal fossa (Figures 1 and 2).
DIAGNOSIS
Congenital adrenal hyperplasia secondary to 3-beta hydroxysteroid dehydrogenase deficiency
DISCUSSION
Various enzyme deficiencies are responsible for congenital adrenal hyperplasia (CAH) with 21-hydroxylase deficiency being the most frequent culprit.1 Multiple subtypes of CAH can lead to bilateral adrenomegaly, evident on imaging which can aid in the diagnosis of unknown or untreated cases.2 In asymptomatic or subclinical patients, the resulting adrenomegaly can predispose patients to adrenal hemorrhage or Waterhouse-Friderichsen Syndrome.3
Three-beta hydroxy-dehydrogenase (3-BHSD) deficiency is extremely rare with approximately 60 affected individuals reported.1 The 3-beta hydroxyseteroid dehydrogenase (3-BHSD) enzyme is one of the most proximal enzymes essential for steroidogenesis in both the adrenal cortex and gonads. Complications in males often include hypospadias, ambiguous genitalia, and infertility.1
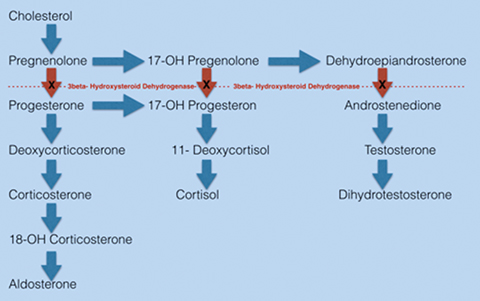
Most patients with 3-BHSD deficiency will require treatment with mineralocorticoid, glucocorticoid and androgen replacement. The level of 3-BHSD enzyme function can vary in patients with deficient enzymatic activity.1 We postulate that our patient has residual enzyme function, as he was non-adherent to medication and asymptomatic for nearly three years. Furthermore, the imaging findings upon presentation likely represent a result of chronic adrenal stimulation and steroid precursor buildup of cholesterol, pregnenolone, 17-OH pregnenolone, and dehydroepiandrosterone (Figure 3).4 The feared complication of CAH is adrenal crisis; a life threatening reaction to low cortisol characterized by hypoglycemia, hyponatremia and hypovolemic shock. Patients with CAH may also struggle with infertility.
CONCLUSION
Imaging findings of adrenomegaly can be seen with 3-BHSD deficiency, a subtype of CAH that leads to deficiencies in mineralocorticoid, glucocorticoid, and androgen synthesis. Although some enzyme function may persist, treatment with fludrocortisone, prednisone, and testosterone is the standard of care. Patients who are withheld treatment risk an adrenal crisis, especially when stressful life events occur.
REFERENCES
- Pang S. Congenital Adrenal Hyperplasia owing to 3 Beta-Hydroxysteroid Dehydrogenase Deficiency. Endocrineo Metab Clin North Am. 2001 Mar; 30(1):81-99, vi-vii. Review
- Harinarayana CV, Renu G, Ammini AC, Khurana ML, Ved P, Karmarkar MG, Ahuja MM, Berry M. Computed tomography in untreated congenital adrenal hyperplasia. Pediatr Radiol. 1991;21(2):103-105.
- Saad AF, Ford KL, Deprisco G, Smerud MJ. Adrenomegaly and septic adrenal hemorrhage (Waterhouse-Friderichsen syndrome) in the setting of adrenal hyperplasia. Proc (Bayl Univ Med Cent). 2013; 26(3): 268-269
- White PC. Neonatal screening for congenital adrenal hyperpasia. Nature Reviews Endocrinology. 2009; 5: 490-498.
Citation
S P, D G, J Y. Congenital adrenal hyperplasia secondary to 3-beta hydroxysteroid dehydrogenase deficiency. Appl Radiol. 2017;(11):46-47.
November 13, 2017