Contrast-enhanced MRI: History and Current Recommendations
Images
Magnetic resonance imaging (MRI) depicts intrinsic contrast between structures using differences in magnetization properties, but it was recognized early in the development of MRI that paramagnetic agents enhance tissue discrimination.1-3 Gadolinium (Gd) was shown to have a particularly strong effect on shortening the T1 and T2 relaxation times of hydrogen protons.4 Notably, many paramagnetic ions are relatively toxic in their natural, free ionic forms, thus chelation is required to reduce toxicity before injection into living organisms.5 When chelated, toxicity is minimized, but T1 and T2 relaxivity, while diminished, are not eliminated.6
In 1984, Schering filed the first patent application on an MRI contrast agent called Gd(III) diethylenetriaminepentaacetate (Gd-DTPA) or gadopentetate dimeglumine. Gd-DTPA, marketed as Magnevist, served as the forefather of Gd-based contrast agents (GBCAs).7 A preclinical study published in 1984 showed that “the combination of strong proton relaxation, in vivo stability, rapid urinary excretion, and high tolerance favors the further development and the potential clinical application of Gd-DTPA as a contrast enhancer in magnetic resonance imaging.”6 The results of this landmark study also helped lay the groundwork for subsequent permutations of chelated agents, making this article the most cited publication in the American Journal of Roentgenology (AJR) at its centennial.8
In 1984, the first images performed with intravenous gadopentetate dimeglumine in patients with cerebral, liver, and bladder tumors were published.5 In 1988, gadopentetate dimeglumine received approval for clinical use in the United States, Germany, and Japan.3 At that time it received US Food and Drug Administration (FDA) approval for contrast-enhanced MRI (CE-MRI) of the central nervous system, an approval that was then extended to the rest of the body (except the heart) five years later.7
Refinement of Gadolinium MRI Contrast
During the decade following FDA approval, gadopentetate dimeglumine use increased dramatically.9 Those developing competitor GBCAs sought to develop improved agents, predominantly with higher relaxivity. Based on ligand structure, GBCAs are divided into two groups, linear and macrocyclic, both of which can be ionic or non-ionic in overall charge. Linear agents have an extended organic molecular ligand that enfolds around the ion, while macrocyclic agents confine the ion in a preformed central cavity.10 Transmetalation, also called dechelation, occurs when competing endogenous metals, including zinc, copper, calcium, and iron, destabilize and thus accelerate dissociation of GBCAs into the Gd ion in vivo.11
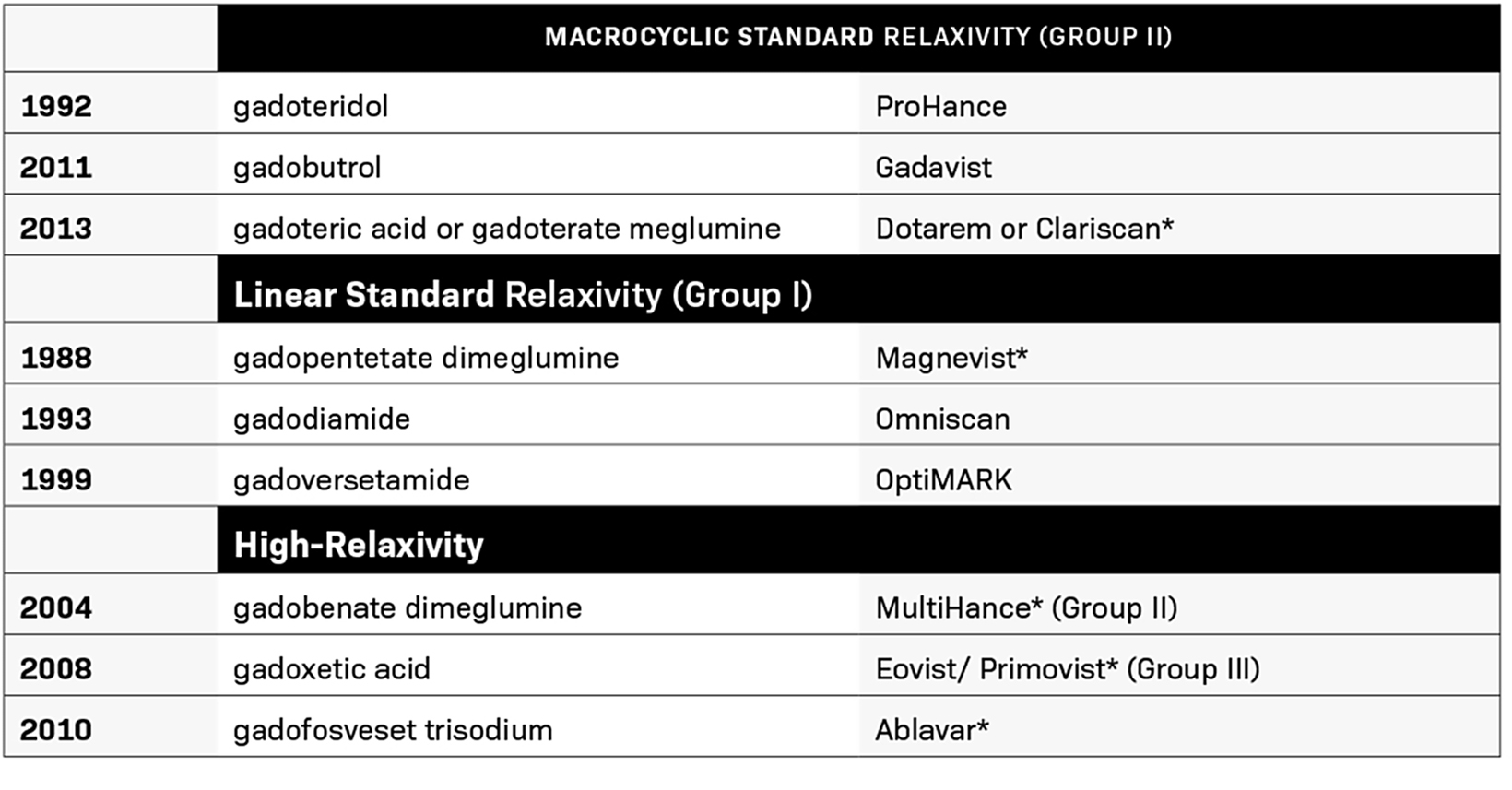
Between 1988 and 2013, the FDA approved nine contrast agents (Table 1). During this time, GBCA use evolved to include higher than standard 0.1 mmol/kg doses, with double doses being used for MR angiography (MRA) and triple doses used for certain applications.12,13 Substantial evidence demonstrated that higher doses provided additional diagnostic yield with few associated safety concerns, and one GBCA, gadoteridol, received an indication for 0.3 mmol/kg; it retains that triple dose indication today.14
Developments in MRI technology and GBCAs increased the value of CE-MRI; hence, the use of GBCAs in approximately 30 million MRI procedures annually.3 More than 450 million doses of GBCAs have been given since its introduction in 1988.15 GBCAs increase tissue differentiation, allowing for evaluation of perfusion as well as the characterization of lesions, and are used particularly for MRA and for MRI of the CNS, abdomen, breast, and heart. From their early days, GBCAs were well tolerated, with a low rate of adverse events.16 Notably, GBCAs were widely perceived as safe alternatives in patients with poor renal function who could not receive iodinated contrast media.17
MRI Contrast Safety: Nephrogenic Systemic Fibrosis (NSF)
In 1997, over a decade after GBCAs were first administered to humans, some renal dialysis patients began developing unexplained skin thickening after unsuccessful renal transplantations.18 Dr Philip LeBoit, a dermopathologist, deemed the disorder “scleromyxedema-like,” owing to the presence of “peau d’orange” skin findings without the IgG lambda paraprotein.19 A collaborative, multicenter, clinicopathological study ensued to determine the cause.
Subsequent studies demonstrating involvement of deeper structures beyond the skin indicated a systemic disease-related etiology;20 the condition, originally named nephrogenic fibrosing dermopathy (NFD) due to its skin manifestations, ultimately was renamed to the more comprehensive nephrogenic systemic fibrosis (NSF).
The first link between NSF and GBCAs was suggested in 2006, when Austria’s Dr Thomas Grobner documented the development of skin symptoms in five patients with end-stage renal disease between two and four weeks after undergoing contrast-enhanced MRA with gadodiamide.21 Subsequent case analyses have demonstrated that most patients who develop NSF do so within three to six months of GBCA exposure.9
Studies suggest NSF results from a chemical transformation of the GBCA molecules, leading to Gd release (dechelation) and subsequent accumulation. The linear GBCAs of the high-risk group are more prone to releasing Gd than the macrocyclic GBCAs of the low-risk group, and more likely to induce NSF. Yet, it is often difficult to attribute individual NSF cases to the administration of a specific GBCA, as most of the patients received multiple GBCAs before diagnosis. By combining both clinical and histopathologic criteria, Girardi, et al, have developed a scoring system that allows the exclusion of conditions mimicking NSF while facilitating its reproducible and accurate diagnosis.22
Understanding NSF is also made more challenging by its rarity, with only 400-800 cases worldwide. Most, but not all, have been associated with GBCAs. In a 2018 evaluation of 145 million administered doses of gadopentetate dimeglumine, only 74 patients had reports diagnostic of or consistent with NSF.9 To evaluate the association of NSF with high-risk agents, Edwards, et al, analyzed three public safety databases, which included the International Centre for Nephrogenic Systemic Fibrosis Registry (ICNSFR), the Food and Drug Administration Adverse Event Reporting System (FAERS), and a legal data set.
Among 382 biopsy-proven NSF cases, Edwards, et al, found 279 unconfounded cases (involving a single GBCA), all of which involved a linear GBCA.23 Bayer Healthcare published a retrospective analysis of their safety database, which confirmed the greater probability of NSF occurrence when using linear GBCAs. Over a 10-year period, 563 of the 779 NSF reports involved gadopentetate dimeglumine, and Endrikat, et al, found that 220 were unconfounded. They also demonstrated that GBCAs with lower market shares and late market introduction are less likely to be associated with NSF in an unconfounded setting.9 A systematic review of published literature by Attari, et al, led to the identification of 639 patients with biopsy-confirmed NSF. Of these, 405 reported the type of GBCA used. The majority of cases occurred with group I agents, and few cases were associated with group II agents.24
The first GBCA received FDA approval in 1988, nine years before NSF was first described in 1997. In May 2007, the FDA required the addition of a black box warning to the labeling of GBCAs stating that patients with severe renal insufficiency who receive GBCAs are at risk for developing NSF. In September 2010, the FDA further required that all GBCA labels emphasize the need to screen patients for renal dysfunction before administration.25 They also decided that group I agents (gadodiamide, gadopentetate dimeglumine, and gadoversetamide) be contraindicated in those patients, since they are associated with a greater risk of NSF than are group II agents (gadobenate dimeglumine, gadobutrol, gadoteric acid, and gadoteridol).26 Following this black box labeling, NSF nearly disappeared.
The Pendulum Swings Back
New cases of NSF were largely eliminated by screening high-risk patients for renal dysfunction, considering alternative examinations, using the lowest effective contrast dose, and using a Group II or III agent with lower NSF risk. Only seven cases of NSF have been reported after 2008.24,27 However, because of radiologist reticence related to NSF, many patients with renal disease have been denied the benefits of CE-MRI.
The French Pro-FINEST study was the first to estimate the incidence of NSF in patients on long-term dialysis. It found that of 287 patients who underwent CE-MRI [the majority (93.4%) received a macrocyclic GBCA, specifically gadoteric acid (88.9%)], 22 reported a dermatological event within four months after the examination, but none of these cases were diagnosed as NSF.28,29
The international SECURE study evaluated the safety profile of gadoteric acid in 35,499 patients, including individuals with moderate (n = 417), severe (n = 58), or end-stage (n = 7) renal insufficiency. None of the patients with renal dysfunction developed NSF or had a suspicion of NSF after a mean follow-up of at least three months. Similar results were obtained from patients with stage 3 to stage 5 chronic kidney disease, who were given gadobenate dimeglumine or gadobutrol, and there were additional similar studies of gadoteric acid.30
Recognizing the lifesaving benefits of CE-MRI and incorporating the findings of these studies, the American College of Radiology (ACR) in 2017 recommended that renal function screening longer be required for Group II agents in both in- and outpatients.31 In 2020, the ACR and the National Kidney Foundation issued a consensus statement that, depending on the clinical indication, the potential harms of delaying or withholding group II or group III GBCAs for MRI in patients with renal dysfunction should be balanced against the risk of NSF.32
MRI Contrast Safety: Gadolinium Retention
Concerns regarding the perceived safety profile of GBCAs arose again in 2014, with observation of an increase in T1 MR signal within the globus pallidus and dentate nucleus on noncontrast scans in patients who had received GBCAs in the past, indicating that the signal seemed to be coming from Gd retained in the brain of these patients. Such findings were seen even with low GBCA doses in patients with normal renal function and in those with an intact blood-brain barrier, indicating that all patients receiving a GBCA are potentially at risk for Gd retention in the brain.33,34 Similar in vitro and in vivo reports demonstrating Gd retention in bone had been published previously.35,36
Using inductively coupled plasma mass spectrometry (ICP-MS), the T1 hyperintense signal seen on noncontrast scans was confirmed to result from the presence of Gd. Gadolinium was found in the brain following administration of all GBCAs, including macrocyclic agents, albeit at lower levels than following linear agents.37 ICP-MS also detected Gd in bone at much higher levels than in brain tissue.
Like findings related to NSF, Gd retention appears to occur more often with linear GBCAs than with macrocyclic agents, presumably because the macrocyclic GBCAs are more stable and thus hold the toxic Gd ion more tightly, undergoing dechelation less readily.38 Among macrocyclic GBCAs, visible hypersignal thus far has been seen only following high doses of the macrocyclic gadobutrol.39,40
Among linear GBCAs, Gd seems to remain in the body longer after gadodiamide or gadoversetamide administration than after the protein bound gadoxetic acid or gadobenate dimeglumine.41 Whether a linear agent is ionic or nonionic seems also to have an impact; after 15 days, release of the free Gd ion from the nonionic linear GBCAs is about 10 times higher than from the ionic linear GBCAs.42
In response to the T1 signal seen in the brain on noncontrast scans, the FDA released a safety alert in 2015, stating that the agency was “investigating the risk of brain deposits following repeated use of GBCAs for MRI,” owing to reports in the medical literature that patients who underwent four or more contrast MRI scans had enduring GBCA brain deposits “long after the last administration.”43 In 2017, the European Commission suspended marketing of intravenous linear products, including gadodiamide, gadopentetate, dimeglumine, gadoversetamide, and gadobenate dimeglumine (except if used for liver scans).44
That same year, the FDA issued a Safety Announcement requiring that outpatients be given a medication guide in accordance with a new class warning for GBCAs.41 The medication guides alluded to NSF even for Group II agents,45 despite numerous studies that indicated new cases of NSF had been largely eliminated. Further, the medication guides stated that renal function would be screened prior to Group II administration, which would not necessarily be performed under the recommended parameters of the ACR.31,41
Altogether, the Gd retention studies demonstrate that GBCAs have access to various body compartments, including the brain, even in subjects without severe renal dysfunction. GBCAs are cleared from these compartments at different speeds, and they may be partially retained in the tissues for weeks, months, or years, depending not only on their in vivo stability but also on the frequency of administration. The presence of chelated Gd several weeks or even months after injection may reflect a slow but physiological process of washout. To date, the chemical structure of the retained Gd, the clearance rates, and the clinical consequences of this accumulation are unknown.46 Further, while Gd deposition may be dose dependent, to date no reports have suggested neurotoxicity.31
Clinical symptoms reported by patients who believe they have suffered from Gd toxicity and/or retention are mostly nonspecific. Burke, et al, published results of an anonymous survey of 50 patients finding that symptoms occurred either immediately (66%) or within 6 weeks (32%) of GBCA administration. The most common were head/neck symptoms (77.6%), including headache and vision and hearing changes, as well as bone/joint pain (77.6%) and skin changes (59.2%).47 The symptoms overlap somewhat with those of NSF, but their description is based solely on survey data, which creates significant inherent biases.
In 2018, the National Institute of Biomedical Imaging and Bioengineering (NIBIB) assembled an international meeting co-sponsored by the RSNA, ACR, and NIH.46 At the time of that meeting, millions of GBCA doses had been administered and 139 patients with normal or near-normal renal function had been reported to the FDA with non-allergic-like symptoms (eg,. joint pain, fatigue, and cognitive changes) and signs they attributed to GBCAs.46 There is insufficient data to confirm these symptoms were the result of GBCA exposure or Gd retention. Research is ongoing, and the unknowns call for more systematic research; however, with no conclusive evidence of clinical sequelae from retained Gd, assessing the true risk of Gd retention is complicated.
Current Recommendations
Although the clinical implications of Gd retention are unknown as of 2021, it is known that practical applications of GBCAs provide crucial, life-saving medical information.31 While physicians should minimize repeated GBCA administrations when possible, they should also be wary of avoiding or deferring necessary CE-MRI. Balancing risk is important; just as the risk of NSF exists, there is also the potential harm of withholding GBCAs in patients who warrant them. 2021 ACR recommendations no longer call for renal function screening for Group II agents in either inpatients or outpatients.48 The European Medicines Agency position differs slightly from the FDA position, with restriction and removal of linear agents from the market, leaving only macrocyclic GBCAs available for general use. The FDA recommends considering retention characteristics when choosing a GBCA for patients who may be at higher risk for Gd retention, including those who require multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions.10
References
- Wahsner J, Gale EM, Rodríguez-Rodríguez A, Caravan P. Chemistry of MRI contrast agents: current challenges and new frontiers. Chem Rev. 2019;119(2):957-1057.
- Runge VM, Stewart RG, Clanton JA, et al. Work in progress: potential oral and intravenous paramagnetic NMR contrast agents. Radiology. 1983;147(3):789-791.
- Lohrke J, Frenzel T, Endrikat J, et al. 25 years of contrast-enhanced MRI: developments, current challenges and future perspectives. Adv Ther. 2016;33(1):1-28.
- Wesbey GE, Higgins CB, McNamara MT, et al. Effect of gadolinium-DTPA on the magnetic relaxation times of normal and infarcted myocardium. Radiology. 1984;153(1):165-169.
- Carr DH, Brown J, Bydder GM, et al. Gadolinium-DTPA as a contrast agent in MRI: initial clinical experience in 20 patients. AJR Am J Roentgenol. 1984;143(2):215-224.
- Weinmann HJ, Brasch RC, Press WR, Wesbey GE. Characteristics of gadolinium-DTPA complex: a potential NMR contrast agent. AJR Am J Roentgenol. 1984;142(3):619-624.
- Pierre VC, Allen MJ, Caravan P. Contrast agents for MRI: 30+ years and where are we going? J Biol Inorg Chem. 2014;19(2):127-131.
- Zamora CA, Castillo M. Update on imaging contrast agents. Magnetic resonance imaging clinics of North America. 2017;25(4):i-i.
- Endrikat J, Dohanish S, Schleyer N, Schwenke S, Agarwal S, Balzer T. 10 years of nephrogenic systemic fibrosis: a comprehensive analysis of nephrogenic systemic fibrosis reports received by a pharmaceutical company from 2006 to 2016. Invest Radiol. 2018;53(9):541-550.
- Cowling T FN. Macrocyclic and Linear Gadolinium Based Contrast Agents for Adults Undergoing Magnetic Resonance Imaging: A Review of Safety. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2019.
- Tweedle MF, Kanal, Emanuel, Muller, Robert. Considerations in the selection of a new gadolinium-based contrast agent. https://appliedradiology.com/articles/consider
- ations-in-the-selection-of-a-new-gadolinium-based-contrast-agent. Published 2014. Accessed Jul 14 2021.
- Filippi M, Campi A, Martinelli V, Pereira C, Scotti G, Comi G. Transitional progressive multiple sclerosis: MRI and MTI findings. Acta Neurol Scand. 1995;92(2):178-182.
- Essig M, Weber MA, von Tengg-Kobligk H, Knopp MV, Yuh WT, Giesel FL. Contrast-enhanced magnetic resonance imaging of central nervous system tumors: agents, mechanisms, and applications. Top Magn Reson Imaging. 2006;17(2):89-106.
- Use of ProHance® (Gadoteridol): A safe, effective, and versatile contrast agent for MR imaging. https://appliedradiology.com/communities/mr-community/use-of-prohance-gadoteridol-a-safe-effective-and-versatile-contrast-agent-for-mr-imaging. Accessed Jul 14 2021.
- Soloff EV, Wang CL. Safety of gadolinium use in patients with advanced CKD/dialysis: a radiologist’s perspective. Kidney360. 2020:10.34067/KID.0000502019.
- Beckett KR, Moriarity AK, Langer JM. Safe use of contrast media: What the radiologist needs to know. Radiographics. 2015;35(6):1738-1750.
- Ose K, Doue T, Zen K, et al. ‘Gadolinium’ as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69(4):507-509.
- LeBoit PE. What nephrogenic fibrosing dermopathy might be. Arch Dermatol. 2003;139(7):928-930.
- Centers for Disease Control and Prevention (CDC). Fibrosing skin condition among patients with renal disease—United States and Europe, 1997-2002. MMWR Morb Mortal Wkly Rep. 2002 Jan 18;51(2):25-6. PMID: 11820525. JAMA. 2002;287(7):838-838.
- Cowper SE. Nephrogenic systemic fibrosis: an overview. J Am Coll Radiol. C2008;5(1):23-28.
- Grobner T. Gadolinium--a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
- Girardi M, Kay J, Elston DM, Leboit PE, Abu-Alfa A, Cowper SE. Nephrogenic systemic fibrosis: clinicopathological definition and workup recommendations. J Am Acad Dermatol. 2011;65(6):1095-1106.e1097.
- Edwards BJ, Laumann AE, Nardone B, et al. Advancing pharmacovigilance through academic-legal collaboration: the case of gadolinium-based contrast agents and nephrogenic systemic fibrosis-a Research on Adverse Drug Events and Reports (RADAR) report. Br J Radiol. 2014;87(1042):20140307.
- Attari H, Cao Y, Elmholdt TR, Zhao Y, Prince MR. A systematic review of 639 patients with biopsy-confirmed nephrogenic systemic fibrosis. Radiology. 2019;292(2):376-386.
- Yang L, Krefting I, Gorovets A, et al. Nephrogenic systemic fibrosis and class labeling of gadolinium-based contrast agents by the Food and Drug Administration. Radiology. 2012;265(1):248-253.
- Woolen SA, Shankar PR, Gagnier JJ, MacEachern MP, Singer L, Davenport MS. Risk of nephrogenic systemic fibrosis in patients with stage 4 or 5 chronic kidney disease receiving a group II gadolinium-based contrast agent: a systematic review and meta-analysis. JAMA Intern Med. 2020;180(2):223-230.
- Malikova H. Nephrogenic systemic fibrosis: the end of the story? Quant Imaging Med Surg. 2019;9(8):1470-1474.
- Janus N, Launay-Vacher V, Karie S, et al. Prevalence of nephrogenic systemic fibrosis in renal insufficiency patients: results of the FINEST study. Eur J Radiol. 2010;73(2):357-359.
- Amet S, Launay-Vacher V, Clément O, et al. Incidence of nephrogenic systemic fibrosis in patients undergoing dialysis after contrast-enhanced magnetic resonance imaging with gadolinium-based contrast agents: the Prospective Fibrose Nephrogénique Systémique study. Invest Radiol. 2014;49(2):109-115.
- Soyer P, Dohan A, Patkar D, Gottschalk A. Observational study on the safety profile of gadoterate meglumine in 35,499 patients: The SECURE study. J Magn Reson Imaging. 2017;45(4):988-997.
- American College of Radiology (ACR) manual on contrast media. In: Media ACoDaC, ed. 10.3 ed: American College of Radiology; 2017.
- Weinreb JC, Rodby RA, Yee J, et al. Use of intravenous gadolinium-based contrast media in patients with kidney disease: consensus statements from the American College of Radiology and the National Kidney Foundation. Radiology. 2020;298(1):28-35.
- Kanda T, Osawa M, Oba H, et al. High signal intensity in dentate nucleus on unenhanced T1-weighted MR images: association with linear versus macrocyclic gadolinium chelate administration. Radiology. 2015;275(3):803-809.
- McDonald RJ, McDonald JS, Dai D, et al. Comparison of gadolinium concentrations within multiple rat organs after intravenous administration of linear versus macrocyclic gadolinium chelates. Radiology. 2017;285(2):536-545.
- Tweedle MF, Wedeking P, Kumar K. Biodistribution of radiolabeled, formulated gadopentetate, gadoteridol, gadoterate, and gadodiamide in mice and rats. Invest Radiol. 1995;30(6):372-380.
- White GW, Gibby WA, Tweedle MF. Comparison of Gd(DTPA-BMA) (Omniscan) Versus Gd(HP-DO3A) (ProHance) relative to gadolinium retention in human bone tissue by inductively coupled plasma mass spectroscopy. Investigative radiology. 2006;41(3):272-278.
- Murata N, Murata K, Gonzalez-Cuyar LF, Maravilla KR. Gadolinium tissue deposition in brain and bone. Magn Reson Imaging. 2016;34(10):1359-1365.
- Radbruch A, Weberling LD, Kieslich PJ, et al. Gadolinium retention in the dentate nucleus and globus pallidus is dependent on the class of contrast agent. Radiology. 2015;275(3):783-791.
- Robert P, Violas X, Grand S, et al. Linear gadolinium-based contrast agents are associated with brain gadolinium retention in healthy rats. Invest Radiol. 2016;51(2):73-82.
- Bjørnerud A, Vatnehol SAS, Larsson C, Due-Tønnessen P, Hol PK, Groote IR. Signal enhancement of the dentate nucleus at unenhanced MR imaging after very high cumulative doses of the macrocyclic gadolinium-based contrast agent gadobutrol: an observational study. Radiology. 2017;285(2):434-444.
- FDA Drug Safety Communication: FDA warns that gadolinium-based contrast agents (GBCAs) are retained in the body; requires new class warnings. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-gadolinium-based-contrast-agents-gbcas-are-retained-body. Published 2017. Accessed Jul 14 2021.
- Frenzel T, Lengsfeld P, Schirmer H, Hütter J, Weinmann HJ. Stability of gadolinium-based magnetic resonance imaging contrast agents in human serum at 37 degrees C. Invest Radiol. 2008;43(12):817-828.
- FDA Drug Safety Communication: FDA evaluating the risk of brain deposits with repeated use of gadolinium-based contrast agents for magnetic resonance imaging (MRI). https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-evaluating-risk-brain-deposits-repeated-use-gadolinium-based. Published 2017. Accessed Jul 14 2021.
- EMA’s final opinion confirms restrictions on use of linear gadolinium agents in body scans. https://www.ema.europa.eu/en/medicines/human/referrals/gadolinium-containing-contrast-agents. Published 2017. Accessed August 29, 2021.
- FDA: Gadoteridol Medication Guide. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/020131s035lbl.pdf#page16. Updated December 19, 2020. Accessed August 29, 2021.
- McDonald RJ, Levine D, Weinreb J, et al. Gadolinium retention: a research roadmap from the 2018 NIH/ACR/RSNA Workshop on Gadolinium Chelates. Radiology. 2018;289(2):517-534.
- Burke LM, Ramalho M, AlObaidy M, Chang E, Jay M, Semelka RC. Self-reported gadolinium toxicity: A survey of patients with chronic symptoms. Magn Reson Imaging. 2016;34(8):1078-1080.
- ACR Manual On Contrast Media. https://www.acr.org/-/media/ACR/Files/Clinical-Resources/Contrast_Media.pdf. Published 2021. Accessed August 29, 2021
.
References
Citation
LE M, R P, KK P. Contrast-enhanced MRI: History and Current Recommendations. Appl Radiol. 2021;(6):15-19.
November 6, 2021