Fetal MRI of common non-CNS abnormalities: A review
Images
Dr. Victoria is an Assistant Professor of Radiology; Dr. Johnson is Director of Body MR and an Assistant Professor of Clinical Radiology; Dr. Chauvin is an Assistant Professor of Radiology; Dr. Kramer is a Professor Emeritus of Radiology; and Dr. Epelman is an Assistant Professor of Radiology, Children’s Hospital of Philadelphia, Philadelphia, PA.
Given its proven utility, widespread availability, and relatively low cost, sonography is the main imaging tool in obstetrics imaging. However, the modality has limitations, which include lack of an acoustic window in the fetus with oligohydramnios, a small field of view, limited soft-tissue acoustic contrast, and beam attenuation by maternal adipose tissue.1
Over the past decade, fetal magnetic resonance imaging (MRI) has emerged as a powerful adjunct in evaluation of the fetus, affording superior tissue contrast, a large field of view, and relative operator independence. The effective use of this imaging tool in the evaluation of the fetus with non-Central Nervous System (CNS) congenital abnormalities of the body, including the thorax and the gastrointestinal (GI) and genitourinary (GU) systems, is reason for adopting fetal MRI as an adjunct in obstetric imaging.
Technique of fetal MRI
The pregnant patient is usually scanned in the supine position, but a left lateral decubitus position may be helpful during late pregnancy to avoid maternal inferior vena cava (IVC) compression from the gravid uterus. A body array coil is wrapped around the maternal abdomen to improve spatial resolution. Maternal claustrophobia can be alleviated by having a partner hold the patient’s hand during scanning, or by having the patient talk to the technologist and/or partner during the scan.
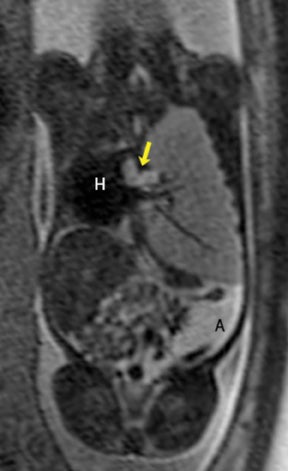
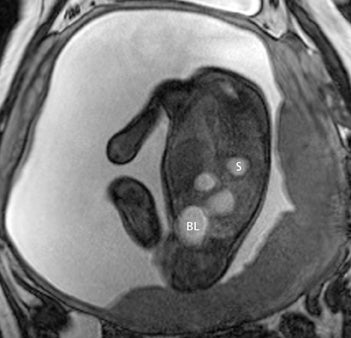
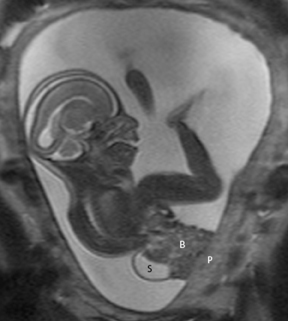
In our practice, the first 2 sequences obtained are orthogonal sagittal and coronal images with respect to the mother, which provide an overall view of the fetus and allow the interpreting radiologist to evaluate fetal situs. The cervix is usually included in these 2 sequences to evaluate for length and competence. The remaining sequences are prescribed with respect to the fetus. Sequences utilized include the T2-weighted half-Fourier acquisition single-shot turbo spin echo (HASTE); the T2W steady-state free procession (TruFISP); echo planar imaging (EPI); and T1W fast low-angle shot (FLASH). HASTE, a black-blood sequence, is the workhorse of fetal imaging, resulting in fine anatomic detail. TruFISP is a white-blood sequence that provides an alternative way of looking at fetal anatomy. EPI is helpful in evaluating the presence of calcifications and blood products. FLASH sequences permit evaluation of fetal structures with normally high T1 signal, including the thyroid, liver, and meconium. Hemorrhage (placental, adrenal, etc.) is suspected when T1W bright signal is seen in these anatomic locations (Figure 1). Evaluation of fetal goiters, although rare, can also be done through T1 FLASH sequences (Figure 2).
Thoracic indications
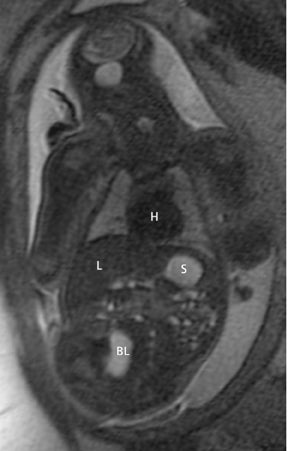
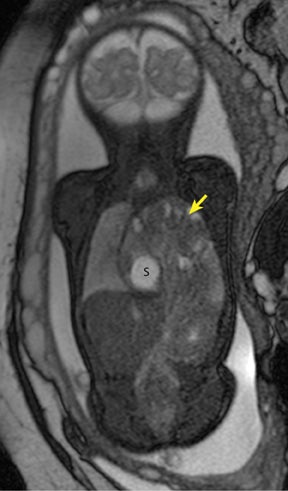
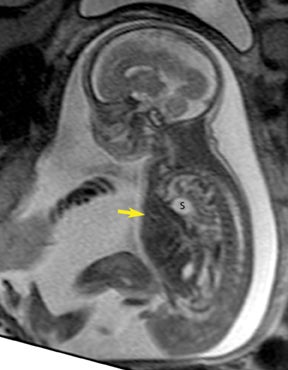
The fetal lung contains a significant amount of alveolar fluid, which results in homogeneously hyperintense T2 signal with respect to the surrounding chest wall, heart, and mediastinal contents in HASTE sequences (Figure 3).2 The thymus has intermediate signal relative to lung; the trachea and bronchi can be visualized due to their fluid content, yielding bright signal on T2-weighted images. T1 images are helpful to evaluate the location of the liver and bowel in the fetus with congenital diaphragmatic hernia.
Congenital lung malformations are rare entities that involve the lung parenchyma and bronchi, as well as their arterial and venous supplies. For the purpose of this review, we will focus on the most commonly encountered lesions in our practice, which include cystic adenomatoid malformation, bronchopulmonary sequestration (BPS), and bronchial atresia (BA). These entities may occur in isolation or as hybrid forms, in which a combination of the different pathologies is present in a single lesion (Figure 4).
Congenital cystic adenomatoid malformations
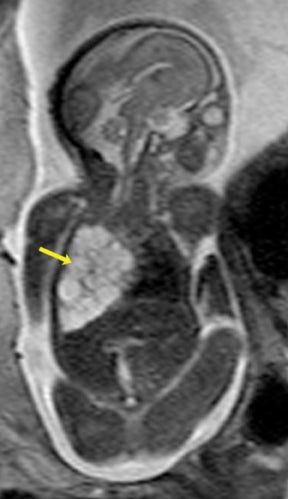
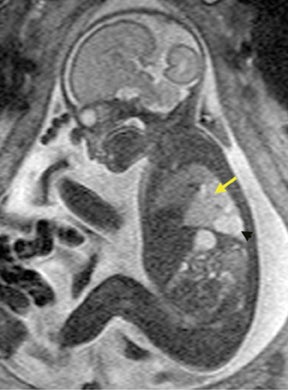
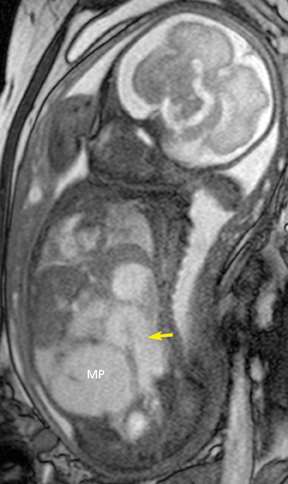
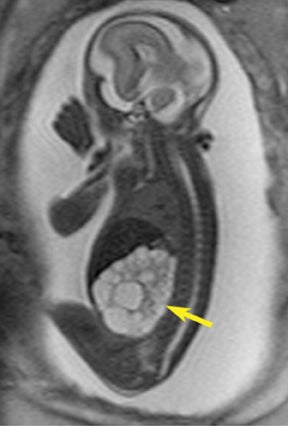
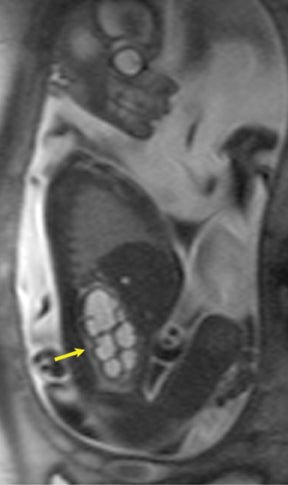
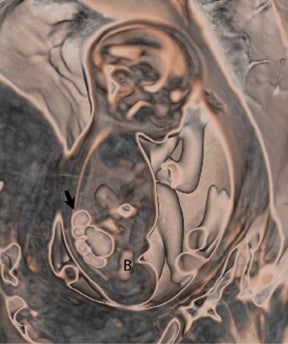
Congenital cystic adenomatoid malformations (CCAM) are the most commonly diagnosed prenatal lung malformations and account for approximately 30% to 40% of all congenital anomalies.2 They are characterized by an abnormal branching of immature bronchioles and a lack of alveolar development, resulting in a mass that may contain both cystic and solid components. CCAMs communicate with the tracheobronchial tree. There are several postnatal CCAM classifications based on pathology;3,4 however, in our practice, prenatal CCAMs are practically classified as containing virtually imperceptible cysts, microcystic CCAM (Figure 5A) or those containing larger cysts, macrocystic CCAM (Figures 5B and 5C). The natural history and prognosis of prenatal CCAMs depend on the size of the malformation, the degree of pulmonary hypoplasia, and the exertion of mass effect manifested by mediastinal shift, cardiac compression, eversion of the hemidiaphragm, and presence of fetal hydrops. In many cases, the malformation progressively shrinks during gestation. In some cases, however, the malformations grow and result in marked mass effect and ominous development of fetal hydrops. The presence of hydrops is nearly uniformly fatal without intervention and is an indication for fetal surgery.5 Fetal surgery may range from open resection for large microcystic lesions to thoracoamniotic shunt placement for the macrocystic lesion with a dominant cyst. By MR, CCAMs present prenatally as homogeneous or heterogeneous solid or cystic masses with blood usually supplied by the pulmonary circulation.
Bronchopulmonary sequestrations
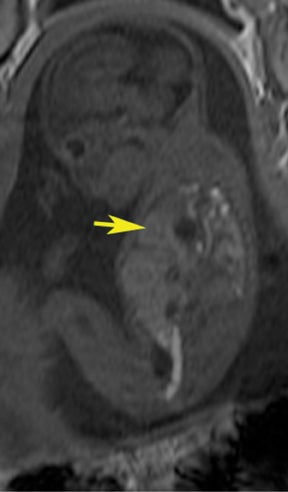
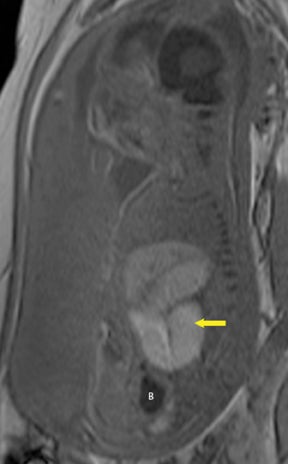
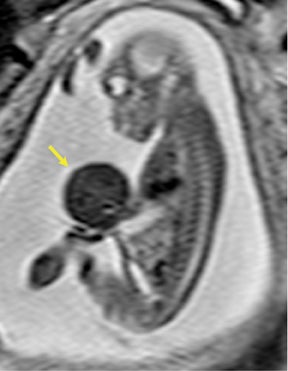
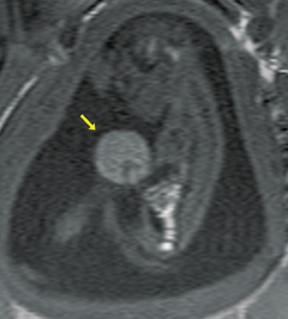
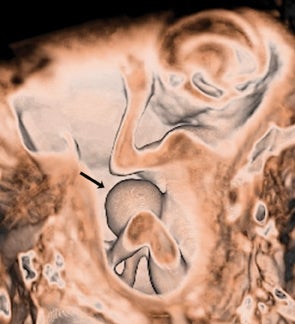
Bronchopulmonary sequestration (BPS) involves abnormal, nonfunctioning pulmonary tissue that does not connect with the normal tracheobronchial tree and receives its vascular supply from the systemic circulation. These malformations are classified as intra- and extralobar. Intralobar sequestrations are the most common, accounting for about 75% of cases, and are characterized by being enveloped by visceral lung pleura. Extralobar sequestrations have their own pleura and may be found in a supra- or infradiaphragmatic location. Both types of sequestrations are more commonly found in the lower lobes; the left is more common than the right. By MR, the characteristics of BPSs are similar to CCAMs: solid, T2 hyperintense masses, usually well defined (Figure 6). Differentiation between CCAM and intrathoracic BPS may be challenging, but identification of a systemic arterial supply, most commonly the aorta, indicates a diagnosis of sequestration.
The differential diagnosis of an extralobar subdiaphragmatic BPS includes adrenal hemorrhage (Figure 1) and neuroblastoma (Figure 7); sequestrations occur more often on the left and present as solid masses with systemic blood supply, whereas neuroblastomas occur more often on the right side. Adrenal hemorrhages may demonstrate T1 hyperintensity suggestive of blood products.
Bronchial atresia
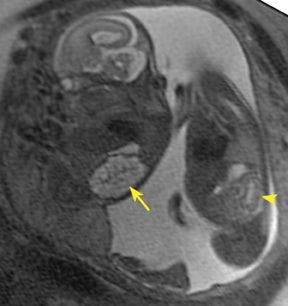
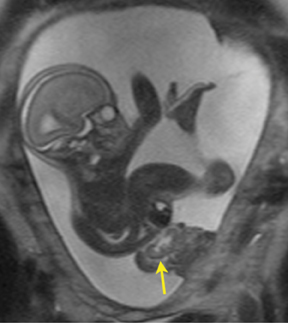
Bronchial atresia (BA) is characterized by atresia or stenosis of a lobar, segmental or subsegmental bronchus at or near its origin, resulting in a blind-ending atretic proximal bronchus and a distended, fluid- filled distal airway that contains variable amounts of mucus, also called mucocele.6 In the prenatal period, the lung distal to the mucocele, although histologically normal in architecture, becomes fluid filled as the fetal lung fluid cannot exit through the obstructed bronchus. The retained fluid results in a T2 hyperintense distal solid-appearing mass, which is the distended, fluid-filled lung (Figure 8). Upper lobe bronchi are more commonly affected; middle and lower lobes are rarely affected. By MR, the fetal appearance of BA is that of a T2 hyperintense mass with a central cyst (mucocele) or centrally dilated bronchi (bronchocele). As with the other pulmonary lesions, mass effect resulting in hemodynamic compromise of the fetus and hydrops should be carefully monitored.
Foregut duplication cysts
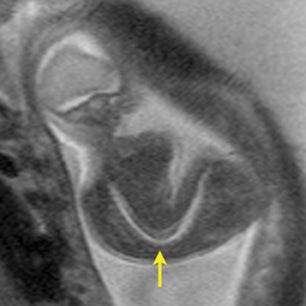
Intrathoracic foregut duplication cysts are the result of developmental malformation of the embryonic foregut. They are divided into bronchogenic, esophageal duplication, and neuroenteric cysts.7 Bronchogenic cysts reflect abnormal budding and development of the ventral foregut during the first trimester of gestation. They may be found anywhere along the tracheobronchial tree. The diagnosis is confirmed when respiratory epithelium is seen on histologic analysis. Esophageal or enteric duplication cysts result from maldevelopment of the posterior division of the embryonic foregut (Figure 9). Submucosa or muscular layer of the GI tract confirms the diagnosis. Neuroenteric cysts are found in the posterior mediastinum and may communicate with the spinal canal, a condition often associated with underlying bony abnormalities of the spine.
Congenital diaphragmatic hernia
Congenital diaphragmatic hernias (CDH) are potentially life-threatening lesions that result from an anatomical defect in the diaphragm that permits abdominal contents to herniate into the thoracic cavity. It occurs in approximately 1 in 2,000 to 1 in 4,000 live births.8 Left-sided hernias are the most common type, accounting for about 85% of cases; the remaining 15% comprise right-sided and bilateral hernias. CDHs result from the failure of closure of the foramen of Bochdalek between the 8th and 10th week of gestation.5 The cause of this failure is uncertain, but one hypothesis includes a disruption of the normal signaling pathway due to either genetic or environmental triggers.
The postnatal morbidity and mortality of the infant with a CDH is due primarily to the pulmonary hypoplasia and pulmonary hypertension that results from chronic compression by the herniated abdominal contents during intrauterine development. The lungs are histologically abnormal, with fewer alveoli, thickened alveolar walls, and increased interstitial spaces, leading to decreased gas-exchange surface area. The pulmonary vasculature is also abnormal with a decrease in the number of vessels along with thickening and muscularization of the wall. Increased vascular reactivity associated with hypoxia then leads to pulmonary hypertension.9
Definitive diagnosis of a CDH relies on the identification of abdominal contents within the fetal thorax.10 Ultrasound has been the mainstay of prenatal diagnosis of CDH; the sonographic findings are beyond the scope of this review, but described in multiple prior studies.10,11 Liver position is the most significant and reproducible independent determinant of survival with liver herniation into the chest predictive of poor outcome.5,12 A recent series demonstrates the mortality of patients with liver “up” (or above the expected location of the diaphragm) to be as high as 65% compared to 7% when the liver is “down.”10 Yet, the sonographic determination of liver position in the fetus with CDH may be difficult, as the liver and lung have similar echogenicity.
MRI has proven to be a resourceful tool in the obstetrics imaging armamentarium, and it has been shown to provide additional information to that demonstrated by ultrasound (US) in 38% to 50% of fetuses with chest lesions.13 By MRI, identification of the fetal bowel and lung is straightforward. The presence of herniated liver can easily be ascertained by a combination of T1 sequences (the liver would be bright) and T2 (dark signal), as seen in Figure 10.
Another important contribution of fetal MRI in the evaluation of the fetus with CDH is the assessment of fetal lung volumes as an indication of the degree of pulmonary hypoplasia.11 MRI has shown a correlation between the degree of pulmonary hypoplasia, as reflected by the measured MRI lung volume, and postnatal survival.
Fetal MRI of the GI system
Morphological development of the GI tract occurs predominantly during the first trimester, while function begins in the second trimester.14 Meconium is produced after week 13 of gestation from secretions of the liver and intestinal glands, desquamated intestinal epithelium, and some amniotic fluid. It slowly migrates from the small bowel to the rectum with advancing gestation. At the same time, anal sphincter pressure increases, resulting in a functional obstruction of the anal canal at 20 weeks and progressive accumulation of the meconium in the distal large bowel.
By MRI, meconium has T1 bright signal (Figure 3), possibly due to its high protein content or its paramagnetic mineral constituents (iron, copper, and manganese).14 At 20 weeks and beyond, the colon and rectum become a meconium reservoir and demonstrate high T1, low T2 signal. The esophagus, stomach and proximal small bowel, contain swallowed amniotic fluid, resulting in low T1 and high T2 signal. Any variation on this scheme may represent pathology. Below are examples of commonly-encountered fetal GI pathology.
Esophageal atresia
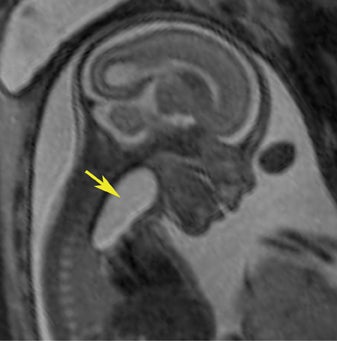
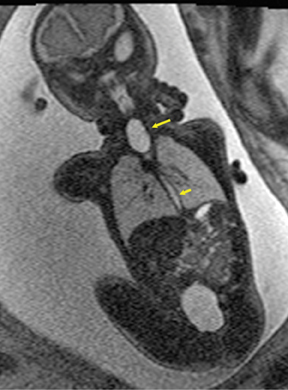
Esophageal atresia with or without distal fistula is a difficult diagnosis to make prenatally and is usually based on indirect findings that include polyhydramnios caused by obstruction of fetal swallowing, a persistently small stomach and possibly, but rarely seen, a dilated upper esophageal pouch (Fig- ure 11). About half of infants with esophageal atresia have other associated anomalies grouped under the acronym of VACTERL (vertebral defects, anal atresia, cardiac anomalies, trachoesophageal fistula, radial defects, renal, and limb anomalies), mandating a careful fetal survey for additional anomalies.
Duodenal atresia
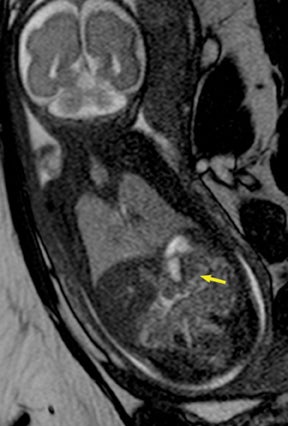
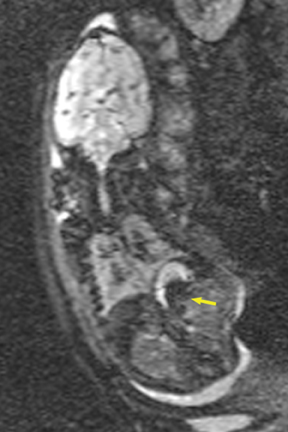
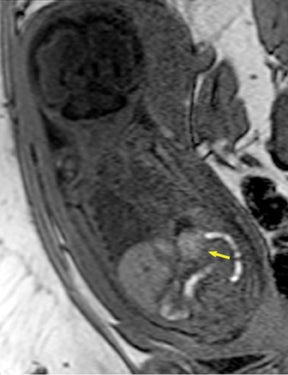
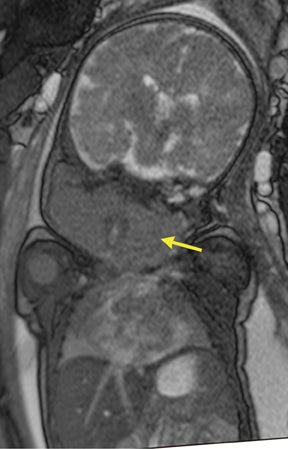
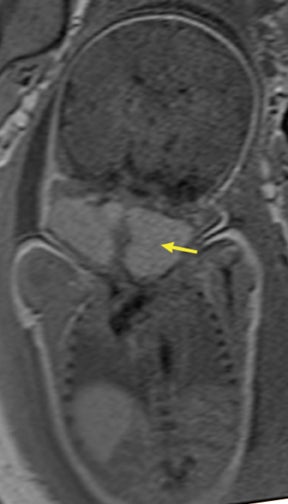
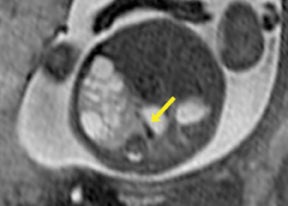
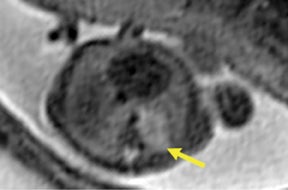
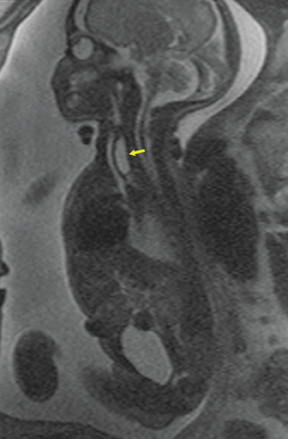
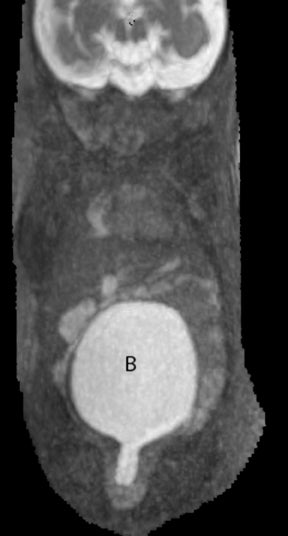
Duodenal obstruction from atresia or stenosis affects 1 in 6,000 to 1 in 10,000 infants.15 This abnormality is thought to be due to lack of recanalization of the duodenal lumen from weeks 8 to 10 of gestation. In more than 50% of cases it is associated with other congenital anomalies, including trisomy 21 (30% to 40%), anorectal malformation, esophageal atresia, and congenital heart disease. The prenatal sonographic findings of duodenal atresia are the classic “double bubbles,” which mirror the MR findings, as seen in Figure 12. The “bubbles” represent the distended stomach and proximal duodenum. Polyhydramnios may be present, as the swallowed fluid cannot enter the proximal small bowel loops.
Small bowel atresia
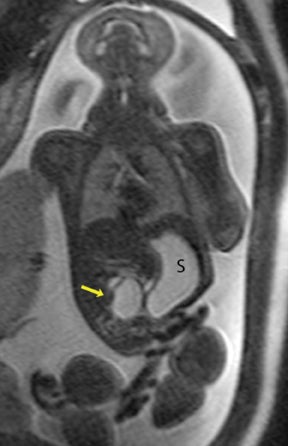
This anomaly occurs in approximately 1 in 3,000 live births (Figure 13). It is thought to be due to the disruption of the mesenteric blood supply to the affected segment occurring after the 12th gestational week. Ischemic necrosis leads to damage of the intestinal segment, possibly leading to fibrous scarring or, in its extreme form, the disappearance of bowel. In the case of the jejunum, atresias tend to be multiple. The jejunum has the capacity to dilate considerably before perforation occurs. This is in contradistinction with ileal atresias, which tend to be solitary, and where perforation may occur with very little dilatation.16 Fetal ascites in the setting of bowel obstruction suggests bowel perforation.
Fetal MRI of the GU system
Genitourinary anomalies account for 14% to 40% of congenital anomalies detected on prenatal US. MRI can be particularly helpful in cases of oligohydramnios, where the acoustic window for US is limited.
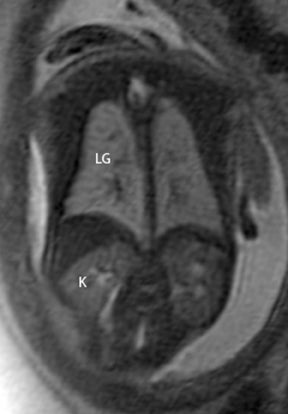
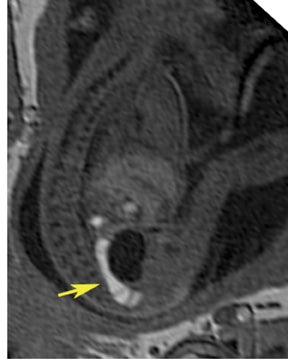
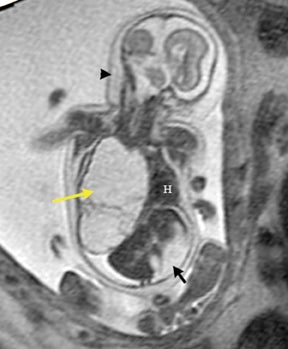
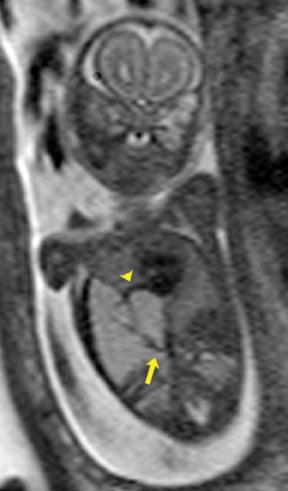
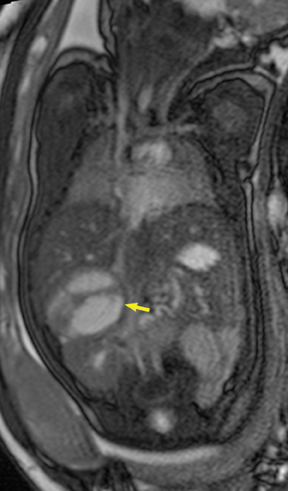
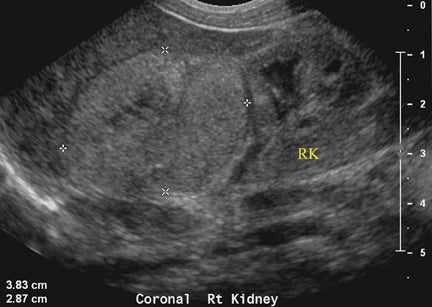
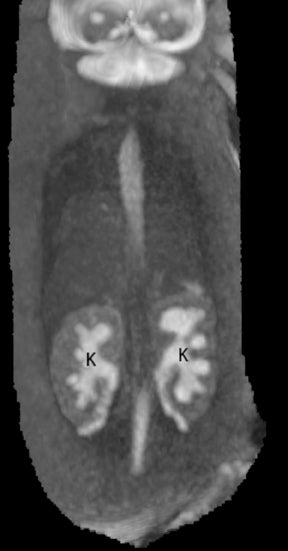
On T2-weighted sequences, the fetal kidneys are seen as ovoid structures with intermediate signal (Figure 3). The presence of T2 bright urine allows the visualization of the collecting system. MRI can help to evaluate the area of abnormality with analysis of the collecting system, ureters, bladder, and urethra. Bilateral versus unilateral involvement is assessed. Thickness of cortex and signal intensity (when compared to maternal kidneys) can help in the evaluation of possible dysplasia. In our practice, common GU indicators for fetal MR include polycystic kidney disease (Figure 14), multicystic dysplastic kidney (Figure 15), ureteropelvic junction (UPJ) obstruction (Figure 16), and posterior urethral valves in the male fetus (Figure 17).
Urinary tract abnormalities may be found in isolation or as part of other anomalies, such as anorectal diseases, congenital heart anomalies, esophageal atresia with or without tracheal fistula, limb abnormalities and neurological disorders. A careful survey of the fetus must be undertaken to assess the possible presence of other abnormalities.
Anterior abdominal wall defects
Gastroschisis and omphaloceles comprise the most common fetal abdominal wall defects. Gastroschisis is defined as a full-thickness paraumbilical defect of the abdominal wall, typically the right side, resulting in prolapse of the bowel, which is then exposed to the amniotic fluid, as there is no membrane covering the defect. It occurs in 1 in 10,000 births, commonly in infants of young mothers. In MR, it is easy to identify the floating loops of bowel in the amniotic fluid. Associated congenital anomalies in the fetus with gastroschisis are rare except those related to other bowel abnormalities, usually intestinal atresia or stenosis from vascular compromise when the blood supply of the eviscerated bowel is compressed at the abdominal wall defect. Thickening or distention of the bowel segments may indicate obstruction/ischemia and are important parameters to report. The condition of the bowel at birth is the single most important prognostic factor in the fetus with gastroschisis.18
Omphalocele
Omphalocele is a midline defect of the abdominal wall at the site of attachment of the umbilical cord that occurs in approximately 1 in 4,000 to 1 in 7,000 births. The extruded organs are covered by parietal peritoneum. Bowel, liver, and stomach may prolapse to the omphalocele sac (Figure 18). Omphaloceles may be found in association with other anomalies in 50% to 80% of cases:19,20 chromosomal abnormalities (trisomy 13 and 18) may occur in 30% to 40% of patients, especially if only small bowel is herniated into the defect; Beckwith-Wiedemann syndrome is found in 5% to 10% of patients with omphalocele.
Additional gastrointestinal anomalies, including intestinal atresias and abnormal fixation of the bowel, are common. Careful fetal survey is warranted to evaluate the coexistence of additional congenital abnormalities.
There are other, rarer types of chest and abdominal wall defects, including those found in the pentalogy of Cantrell, limb-body wall complex (Figure 19), amniotic band syndrome, and bladder and cloacal exstrophy. However, these conditions are beyond the scope of this review.
Conclusion
Superior soft tissue contrast, relative operator independence, and a large field of view are factors that make fetal MRI a great adjunct in obstetric imaging, aiding in the diagnosis of the fetus with congenital abnormalities and in its pre- and postnatal management.
REFERENCES
- Coakley FV, Glenn OA, Qayyum A, et al. Fetal MRI: A developing technique for the developing patient. AJR Am J Roentgenol. 2004;182:243-252.
- Daltro P, Werner H, Gasparetto TD, et al. Congenital chest malformations: A multimodality approach with emphasis on fetal MR imaging. Radiographics. 2010;30:385-395.
- Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8:155-171.
- Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17-37.
- Watanabe M, Flake AW. Fetal surgery: Progress and perspectives. Adv Pediatr. 2010;57:353-372.
- Epelman M, Kreiger PA, Servaes S, et al. Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR. 2010;31:141-157.
- Son JK, Lee EY, Eisenberg RL. Focal nonvascular thoracic masses in children. AJR Am J Roentgenol. 2011;196:W224-W239.
- Gaxiola A, Varon J, Valladolid G. Congenital diaphragmatic hernia: An overview of the etiology and current management. Acta Paediatr. 2009;98:621-627.
- De Buys Roessingh AS, Dinh-Xuan AT. Congenital diaphragmatic hernia: Current status and review of the literature. Eur J Pediatr. 2009;168:393-406.
- Hedrick HL. Management of prenatally diagnosed congenital diaphragmatic hernia. Semin Fetal Neonatal Med. 2010;15:21-27.
- Taylor GA, Atalabi OM, Estroff JA. Imaging of congenital diaphragmatic hernias. Pediatr Radiol. 2009;39:1-16.
- Walsh DS, Hubbard AM, Olutoye OO, et al. Assessment of fetal lung volumes and liver herniation with magnetic resonance imaging in congenital diaphragmatic hernia. Am J Obstet Gynecol. 2000;183:1067-1069.
- Hubbard AM, Adzick NS, Crombleholme TM, et al. Congenital chest lesions: Diagnosis and characterization with prenatal MR imaging. Radiology. 1999;212:43-48.
- Saguintaah M, Couture A, Veyrac C, et al. MRI of the fetal gastrointestinal tract. Pediatr Radiol. 2000;32:395-404.
- Veyrac C, Couture A, Saguintaah M, Baud C. MRI of fetal GI tract abnormalities. Abdom Imaging. 2004;29:411-420.
- Barnewolt CE. Congenital abnormalities of the gastrointestinal tract. Semin Roentgenol. 2004;39:263-281.
- Shinmoto H, Kashima K, Yuasa Y, et al. MR imaging of non-CNS fetal abnormalities: A pictorial essay. Radiographics. 2000;20:1227-1243.
- Durfee SM, Downard CD, Benson CB, Wilson JM. Postnatal outcome of fetuses with the prenatal diagnosis of gastroschisis. J Ultrasound Med. 2002;21:269-274.
- Daltro P, Fricke BL, Kline-Fath BM, et al. Prenatal MRI of congenital abdominal and chest wall defects. AJR Am J Roentgenol. 2005;184: 1010-1016.