Generalized Infantile Myofibromatosis
Images
Case Summary
A newborn presented with respiratory distress and was admitted to the NICU for transient tachypnea of the newborn. Physical examination showed scattered hypopigmented papules and macules along the back, extremities, and buttocks, some of which had central telangiectasia and some of which demonstrated a blue hue. The patient was also noted to have a mass-like deformity of the left upper hip and thigh, and left club foot deformity.
Multiple punch biopsies of the cutaneous lesions showed proliferation of thin-walled vessels, surrounded by small ovoid spindle cells with scant cytoplasm and round nuclei arranged in a lobular pattern, felt to be representative of infantile myofibromas. Biopsy of the left thigh was also consistent with an infantile myofibroma.
Imaging Findings
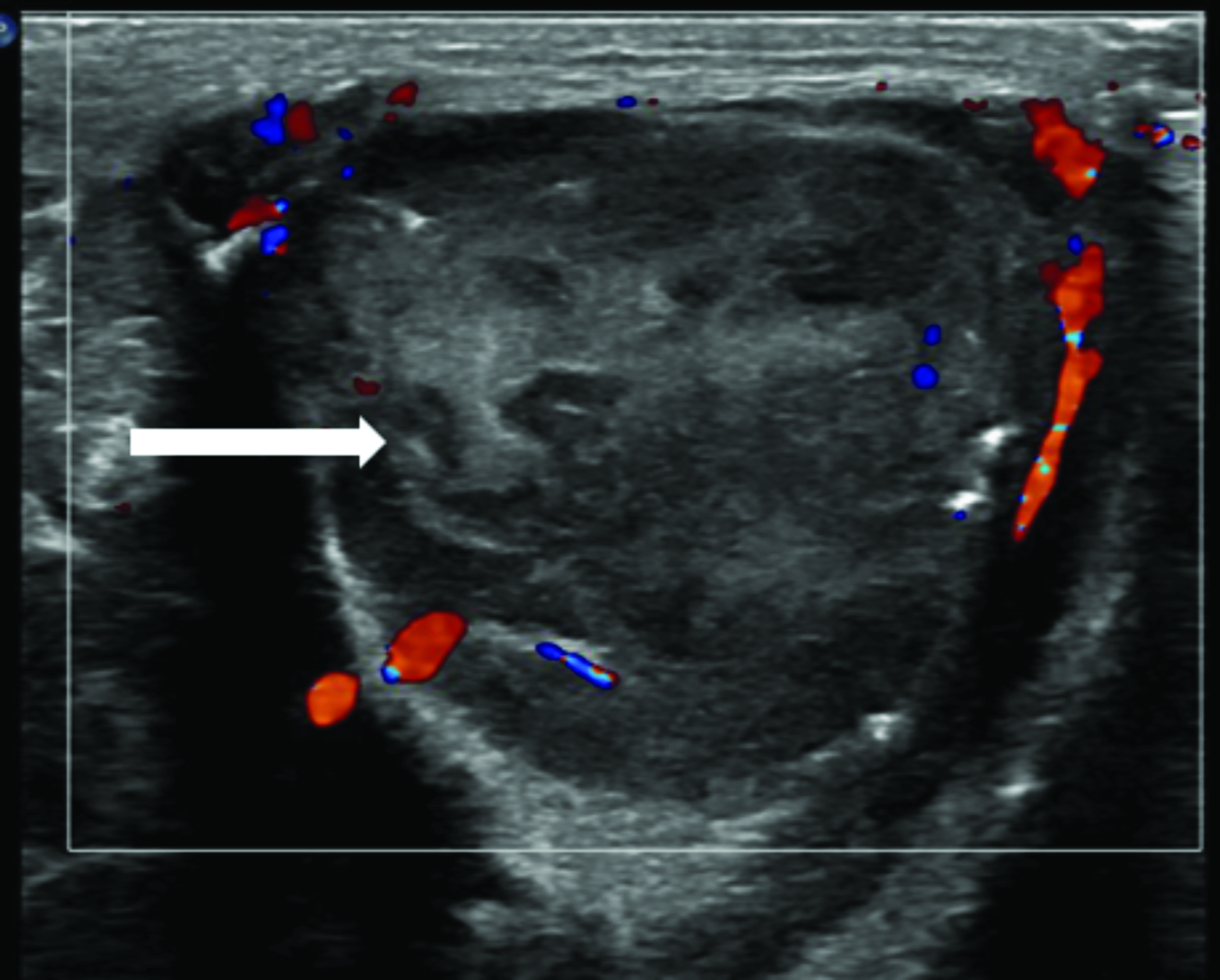
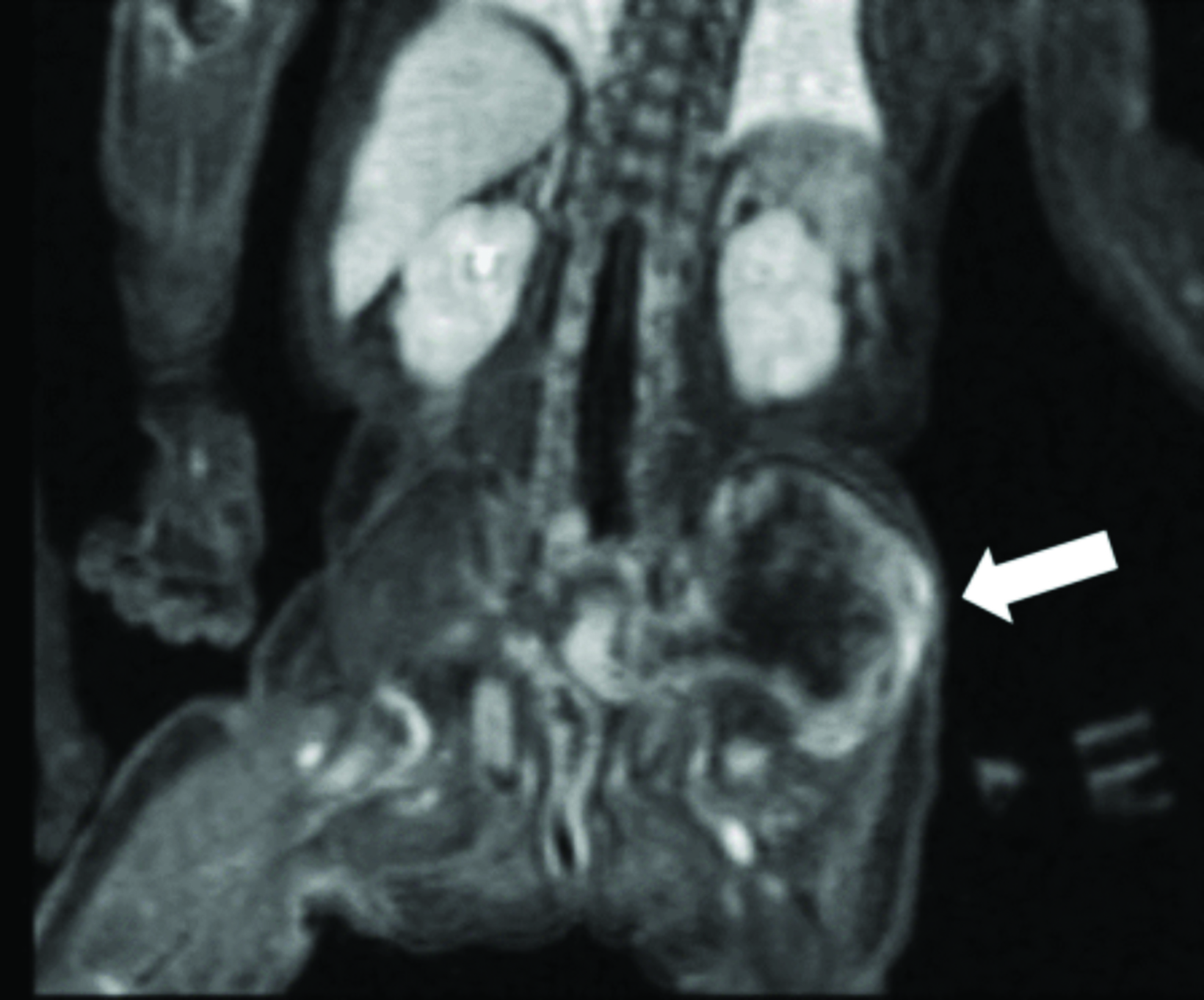
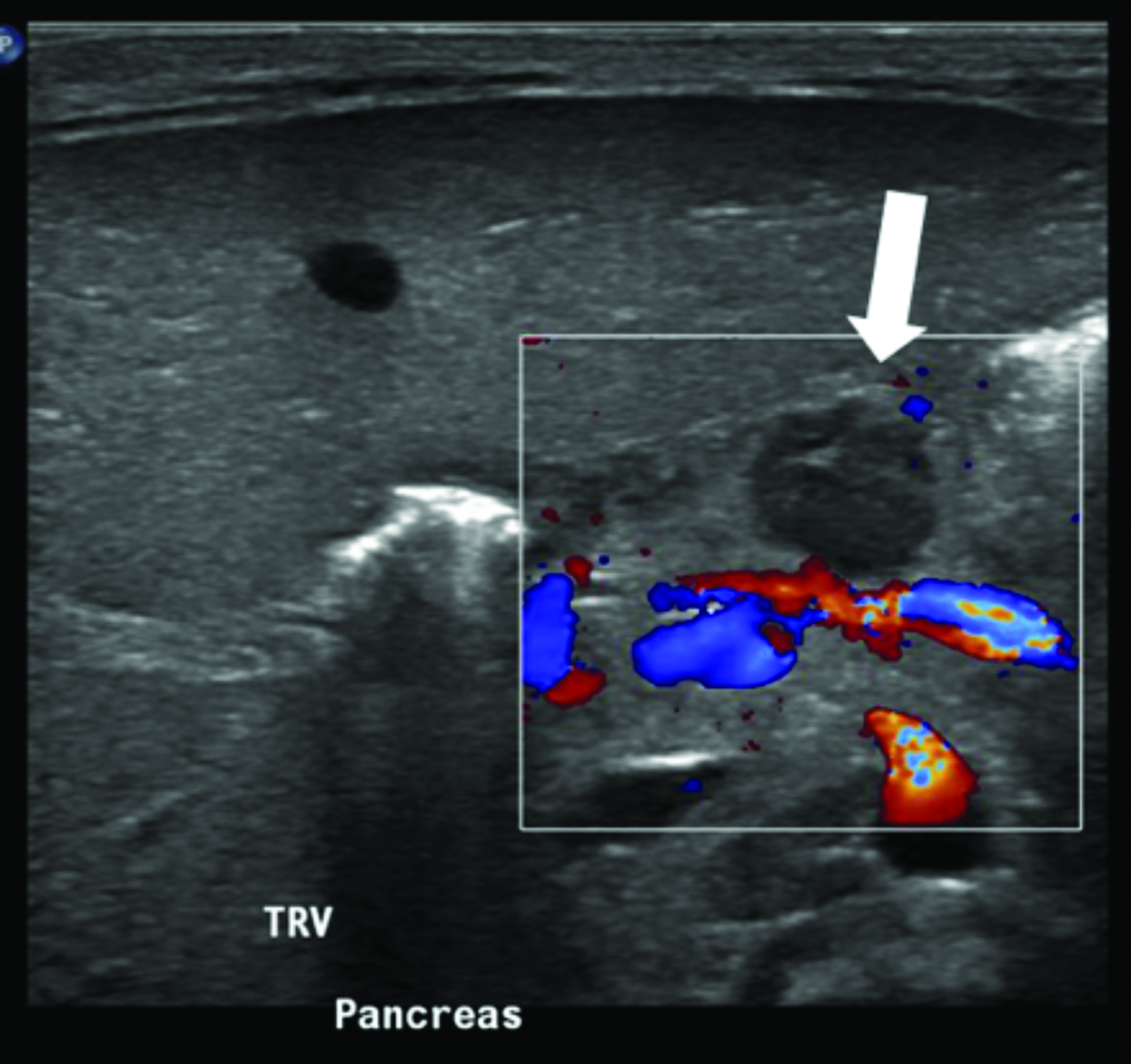
Multimodality imaging demonstrated multiple, well-defined masses disseminated throughout the body. Ultrasound of the left thigh demonstrated a large heterogeneous, hypovascular soft-tissue mass (Figure 1). Magnetic resonance imaging (MRI) of the left thigh/hip showed the mass was predominantly T2 hyperintense, T1 isointense with areas of intrinsic T1 hyperintensity, and demonstrated heterogeneous enhancement on postcontrast T1 imaging (Figure 2). The mass extended into the pelvis. Smaller masses were also seen in the left gluteus muscle and right thigh. Ultrasound of the abdomen showed two heterogeneously echogenic pelvic masses, as well as multiple hypoechoic masses throughout the abdomen, including the retroperitoneum (Figure 3). MR evaluation of the abdomen and pelvis confirmed the ultrasound findings.
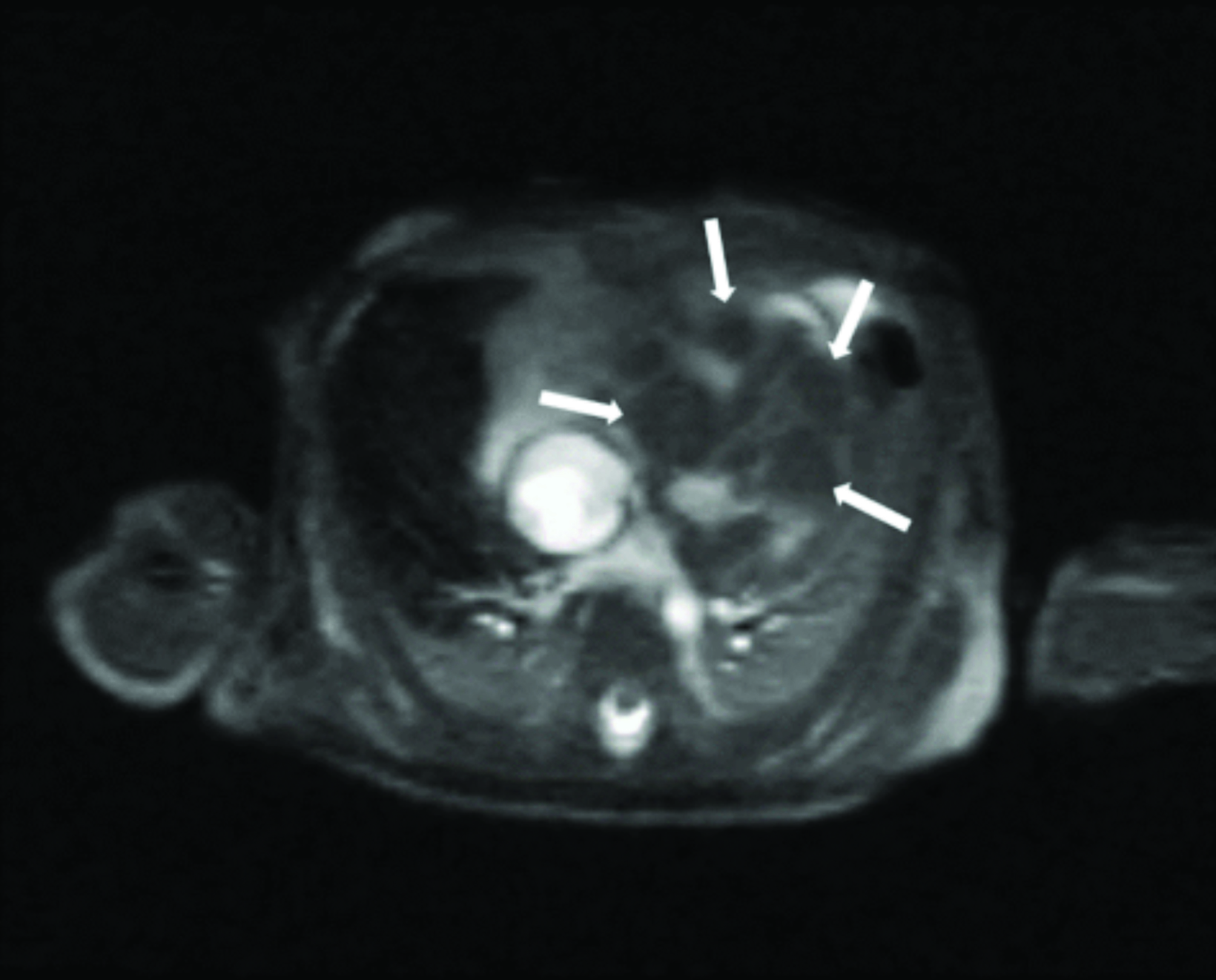
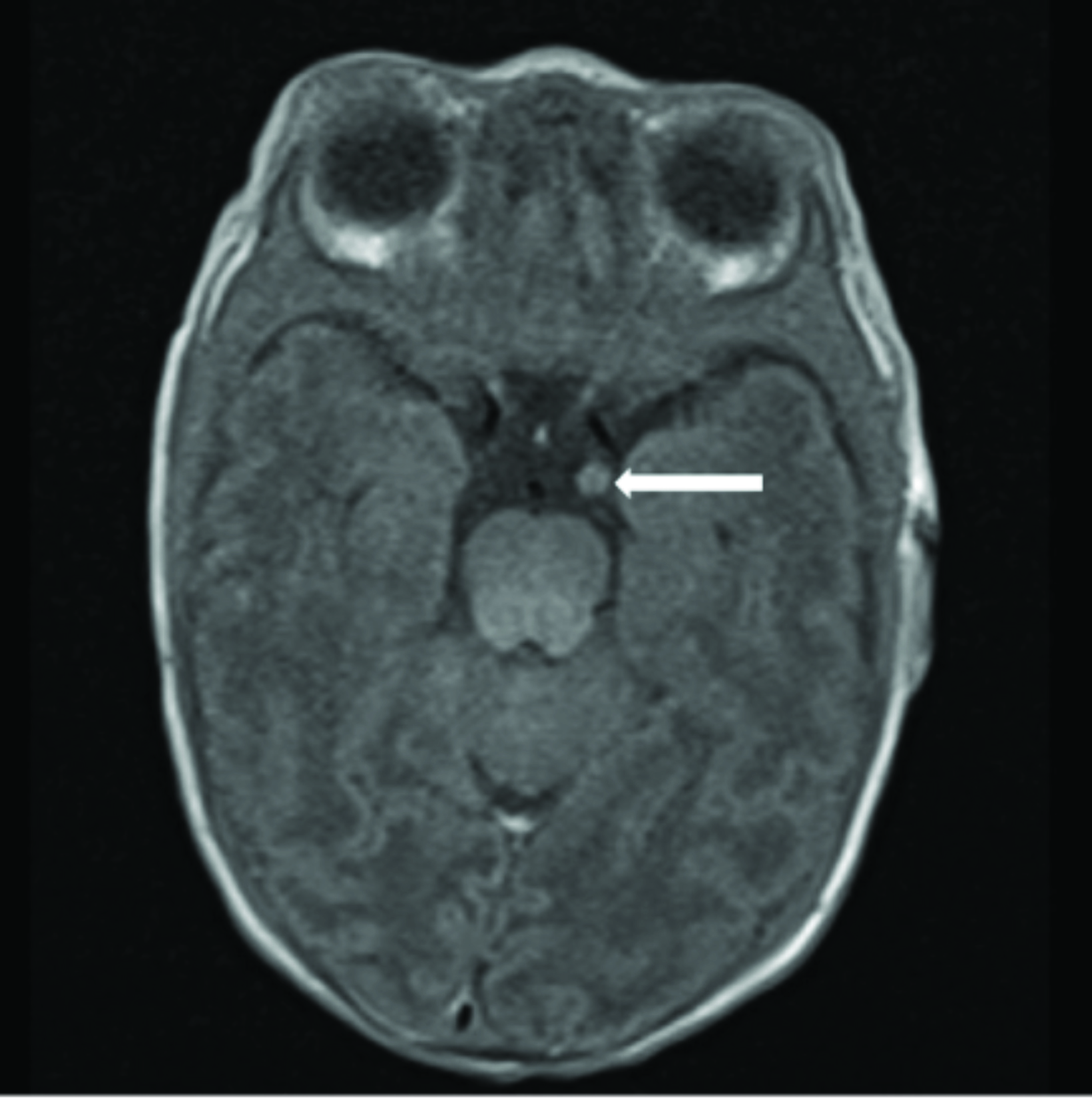
Cardiac MRI revealed multiple round masses in the right and left ventricles, which were T1 isointense and T2 hyperintense (Figure 4). Brain MRI demonstrated a 3.5 mm left supraclinoid soft-tissue mass (Figure 5).
Diagnosis
Generalized infantile myofibromatosis.
Other diagnostic considerations include neonatal rhabdomyosarcoma, infantile fibrosarcoma, neurofibromatosis, and Klippel-Trenaunay syndrome. Neonatal systemic lupus erythematosis is also a consideration, given the cutaneous manifestations.
Discussion
Infantile myofibromatosis is a mesenchymal fibrous tumor of infancy. First described by Stout in 1954, it was named “congenital generalized fibromatosis in the newborn”.1 It was renamed “infantile generalized myofibromatosis” by Chung and Enzinger in 1981 partly because they realized the disorder occurs in both neonates and older infants.2 Although rare, the disorder is one of the most common fibrous tumors in infancy. It typically manifests within the first 24 months of life, and more commonly affects males.3,4 The etiology is unknown; it is theorized that in utero exposure to maternal estrogen may play a role.5
The disorder is classified into three clinical subtypes: solitary, multicentric, and generalized. The solitary form, the most common, manifests with a single firm, painless, skin-colored or reddish-purple cutaneous or subcutaneous nodule.
The multicentric form demonstrates multiple nodules involving the skin, soft tissues, and bones. Both subtypes tend to have a favorable prognosis, with many of the lesions spontaneously regressing within 18 months.3
Generalized myofibromatosis, conversely, portends a worse prognosis, with reported rates of severe morbidity and mortality of up to 75%, predominantly secondary to gastrointestinal (GI) or cardiopulmonary complications.4 The GI tract is the most common site of visceral spread, but pulmonary involvement has the worse prognosis when the viscera is involved.6 Treatment of generalized myofibromatosis varies depending on the severity and extent of disease.
Case reports exist of self-healing generalized myofibromatosis in otherwise healthy infants.6,7 Surgery is usually employed in instances with more extensive involvement or when life-threatening. Chemotherapy (as offered for our patient), and/or radiation therapy is also sometimes used, though chemotherapy has not been shown very effective, as myofibromas are benign tumors with low mitotic activity.3,8
Histologically, myofibromas appear nodular, with short fascicles of spindle-shaped cells along the periphery with elongated nuclei and eosinophilic cytoplasm. Centrally are primitive polygonal or round cells with hyperchromatic nuclei, scant eosinophilic cytoplasm, and indistinct cell margins. Vascular hemangiopericytoma-like proliferations are also sometimes seen.3,5
Initial imaging usually includes CT and ultrasound, although MRI is preferred in suspected generalized myofibromatosis given the modality’s superiority in assessing the viscera and soft tissues. The imaging characteristics of myofibromatosis are nonspecific, and the role of imaging is more to delineate the extent of involvement and less to aid in diagnosis.
On ultrasonography, myofibromas have been described as hypoechoic, mixed echogenicity lesions with decreased flow on color Doppler. CT often shows lesions as isodense to skeletal muscle, sometimes demonstrating mild peripheral enhancement. On MRI, lesions are usually isointense or hypointense to skeletal muscle on T1 imaging, show areas of T2 hyperintensity, and demonstrate predominantly peripheral, heterogeneous enhancement.8,9
Conclusion
Although infantile myofibromatosis is one of the most common fibrous tumors of infancy, the disorder is rare, with a spectrum of presentations and varying prognoses depending on extent of disease and organs involved. In their solitary and multicentric forms, most myofibromas spontaneously resolve and the prognosis is excellent. However, in the generalized form, the prognosis is poor, and treatment options are limited. Imaging findings are nonspecific, and the main role of imaging is to assess for the presence and extent of visceral involvement.
References
- Stout AP. Juvenile fibromatoses. Cancer. 1954;7(5):953-978. https://acsjournals.onlinelibrary.wiley.com/doi/10.1002/1097-0142%28195409%297%3A5%3C953%3A%3AAID- CNCR2820070520%3E3.0.CO%3B2-W.
- Chung E., Enzinger FM. Infantile myofibromatosis. Cancer. 1981;48(8):1807-1818. https://acsjournals.onlinelibrary.wiley.com/doi/10.1002/1097- 0142%2819811015%2948%3A8%3C1807%3A%3AAID-CNCR2820480818%3E3.0.CO%3B2-G.
- Mashiah J, Hadj-Rabia S, Dompmartin A, et al. Infantile myofibromatosis: A series of 28 cases. J Am Acad Dermatol. 2014;71(2):264-270. doi:10.1016/j.jaad.2014.03.035.
- Wiswell TE, Davis J, Cunningham BE, Solenberger R, Thomas PJ. Infantile myofibromatosis: The most common fibrous tumor of infancy. J Pediatr Surg. 1988;23(4):314-318. doi:10.1016/S0022-3468(88)80196-9.
- Oudijk L, den Bakker MA, Hop WCJ, et al. Solitary, multifocal and generalized myofibromas: Clinicopathological and immunohistochemical features of 114 cases. Histopathology. 2012;60(6B):E1-E11. doi:10.1111/j.1365-2559.2012.04221.x
- Martín J, Jordá E, Calduch L, Monteagudo C, Pereda C, G V. Self-healing generalized infantile myofibromatosis. J Eur Acad Dermatology Venereol. 2008;22(2):236-238. doi:10.1111/j.1468-3083.2007.02292.x.
- Léauté-Labrèze C, Labarthe MP, Blanc JF, Sanyas P, Dosquet C, Taïeb A. Self-healing generalized infantile myofibromatosis with elevated urinary bFGF. Pediatr Dermatol. 2001;18(4):305-307. doi:10.1046/j.1525-1470.2001.01933.x.
- Dimson OG, Drolet BA, Southern JF, Rock A, Winthrop AL, Esterly NB. Congenital generalized myofibromatosis in a neonate. Arch Dermatol. 2000;136(5):597-600. doi:10.1001/archderm.136.5.597.
- Naffaa L, Khalifeh I, Salman R, Itani M, Saab R, Al-kutoubi A. Infantile myofibromatosis: review of imaging findings and emphasis on correlation between MRI and histopathological findings. Clin Imaging. 2019;54(March-April 2019):40-47. doi:10.1016/j.clinimag.2018.11.003
References
Citation
TJ B, DA R.Generalized Infantile Myofibromatosis. Appl Radiol. 2022; (3):50-52.
April 26, 2022