Infantile Myofibromatosis: Prenatal and Postnatal Imaging Features
Images
CASE SUMMARY
A 29-year-old 33 weeks and 4 days pregnant underwent a prenatal ultrasound, which demonstrated a dilated bowel loop. A fetal MRI scan was ordered.
IMAGING FINDINGS
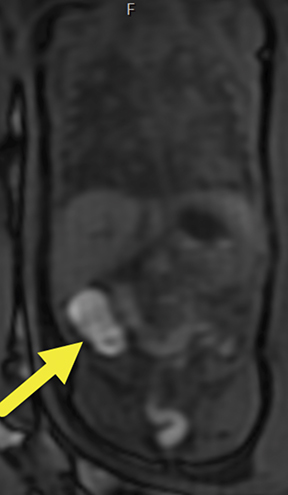
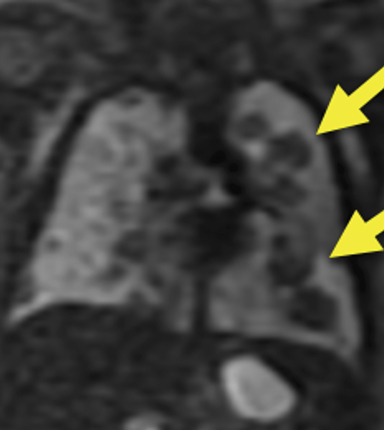
The MRI scan suggested the dilated loop of bowel was colon given the internal high T1 signal compatible with meconium (Figure 1). Additionally, multiple, solid-appearing lung masses were noted, which had not been previously visualized on ultrasound (Figure 2). After thorough review of the MRI images of the entire fetus, no primary lesion was found. The fetal liver, adrenal glands, kidneys, and bladder were all well visualized and normal in appearance. No masses were visualized in the extremities.
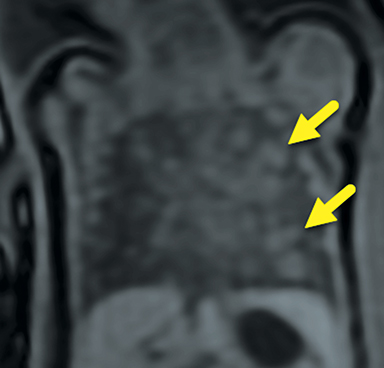
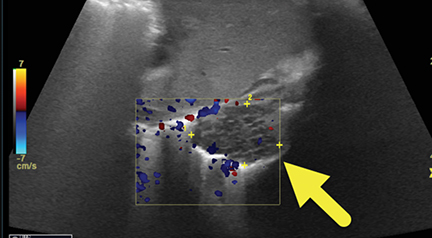
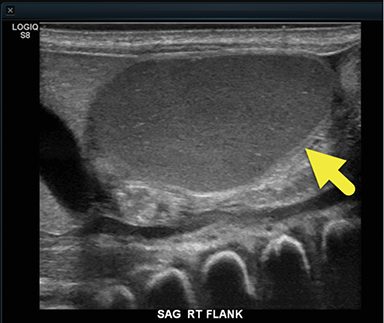
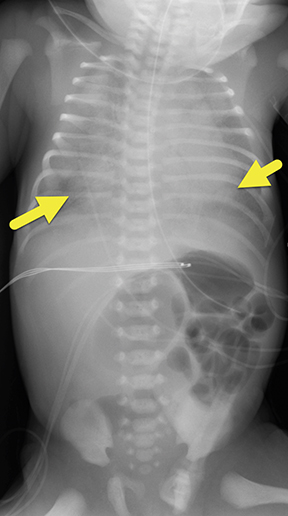
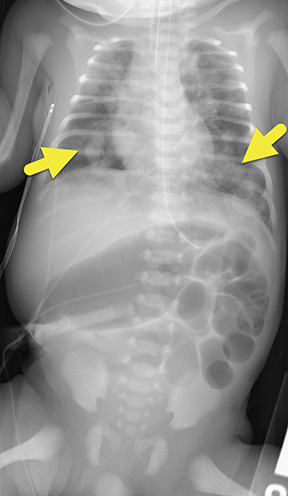
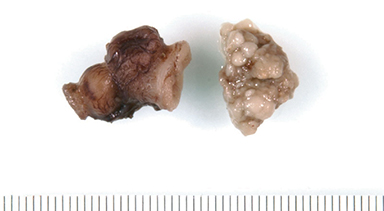
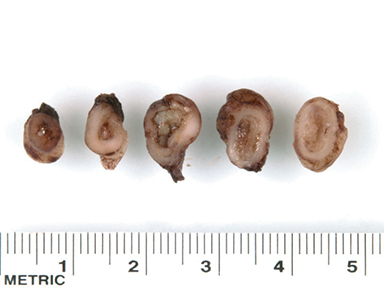
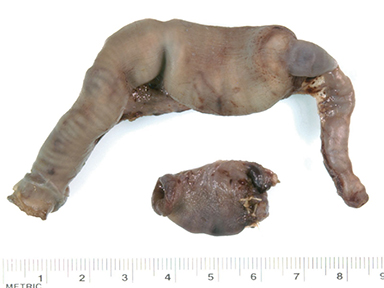
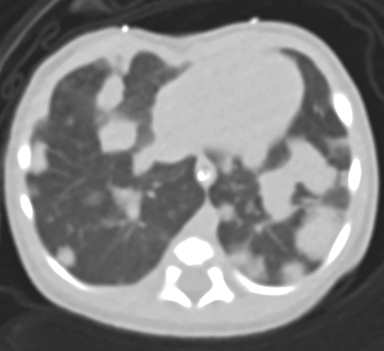
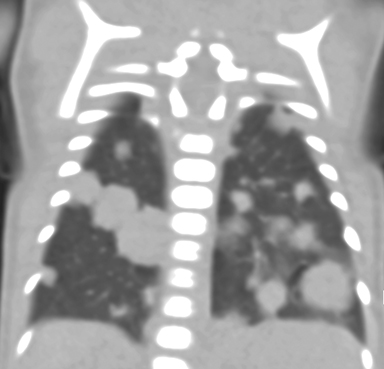
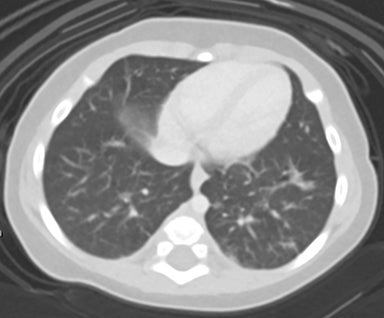
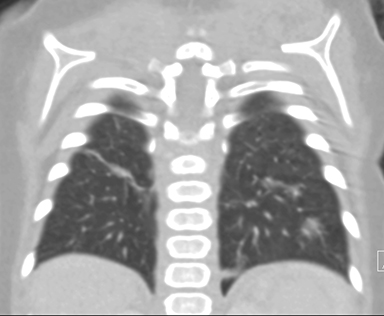
At birth, an abdominal ultrasound scan was performed (Figure 3), which confirmed the prenatal findings. The baby had not passed meconium after 1 day of life; postnatal abdominal radiographs demonstrated a bowel obstruction (Figure 4). The patient then underwent an exploratory laparotomy, which revealed nodules on the bowel serosa, resulting in a bowel obstruction (Figure 5). At histopathologic analysis, these nodules were consistent with infantile myofibromas, with histologic features of smooth muscle cells and fibroblasts. A chest CT scan obtained 5 days after birth confirmed numerous bilateral pulmonary masses previously identified on fetal MRI (Figure 6). The patient also underwent a bone scan at 1.5 weeks of age, which was negative for osseous lesions.
DIAGNOSIS
Infantile myofibromatosis. The differential diagnosis based on the fetal MRI includes metastatic disease from occult malignancy in the mother via transplacental spread (particularly melanoma, breast, and lung cancer); metastatic disease from occult malignancy in the fetus (such as Wilms tumor, neuroblastoma, or rhabdomyosarcoma); or infectious process.
DISCUSSION
The fibromatoses are a diverse group of soft-tissue tumors classified into two major types: superficial and deep. Deep fibromatoses are typically larger, grow more rapidly, and are more aggressive than superficial tumors. Examples include fibromatosis coli, extra-abdominal desmoid tumor, and infantile myofibromatosis.1,2
Infantile myofibromatosis consists of fibrous tumors deposited in the skin, soft tissues, muscles, bones (predominantly metaphyses), and visceral organs (usually the liver and lungs). This entity can be further classified into solitary or multicentric subtypes. The solitary subtype occurs in the skin and subcutaneous tissues, while the multicentric subtype occurs in the skin, subcutaneous tissues, and muscles.2-4
Histopathology is the gold standard for diagnosis. Histologic features include proliferation of myofibroblasts with features of both smooth muscle cells and fibroblasts. Lesions may be well circumscribed or infiltrative.2,3 Ultrasound is often the initial imaging modality for palpable soft tissue lesions. On ultrasound, the lesions appear spherical and hypoechoic, and may demonstrate internal flow on Doppler.2 MRI is the desired imaging modality due to its ability to accurately determine soft tissue extension.3 Typically, lesions are low to intermediate signal on T2 sequences; however, lesions that contain less collagen may demonstrate increased signal.4-7 On CT, lesions are hyperattenuating relative to skeletal muscle and may contain small foci of calcification.1-7
Infantile myofibromatosis can have a variety of imaging features, depending on which visceral organs are affected. Pulmonary lesions can vary on radiography, appearing as reticulonodular opacities or in a pattern suggestive of pulmonary fibrosis. Gastrointestinal involvement may show diffuse narrowing of the affected bowel, with individual myofibromas appearing as multiple filling defects on a barium small-bowel follow-through.1,6 Myofibromas may also affect the bones, including the vertebrae, skull, ribs, pelvis, and particularly the metaphyses of long bones. These lesions appear radiolucent; however, a sclerotic margin may develop over time.1 The cortex may appear expanded, but the periosteum remains intact. These lesions are reported to lack uptake on bone scans, although pathologic fractures may result.3
Although considered controversial, the clinical course is related to visceral organ involvement. Generally, outcomes are worse where the visceral organs are affected.1 However, the extent and locations of myofibromas within the viscera is probably a more important prognostic factor.3 In cases with visceral organ involvement, treatment typically consists of surgical excision of large, solitary lesions.6,7 There is limited success with radiation, steroids, and chemotherapy (methotrexate, vinblastine). In cases without visceral organ involvement usually lesions usually regress spontaneously within 1 or 2 years.4 In this case, owing to visceral organ involvement, chemotherapy was considered; however, it was ultimately decided to observe the patient and treat only if there was radiologic progression and/or clinical deterioration.
CONCLUSION
Infantile myofibromatosis is a solitary or multifocal proliferation of benign, fibrous tumors that can be deposited in the skin, soft tissues, bones, or visceral organs. Despite the presence of visceral organ involvement in our patient, these lesions exhibited spontaneous regression. Although a rare entity, infantile myofibromatosis should be included in the differential diagnosis in a fetus with multiple solid-appearing lung masses. Imaging plays an imperative role in assessing the severity and extent of disease throughout the clinical course .
REFERENCES
- Robbin MR, Murphey MD, Temple HT et-al. Imaging of musculoskeletal fibromatosis. Radiographics. 21 (3): 585-600.
- Murphey MD, Ruble CM, Tyszko SM et-al. From the archives of the AFIP: musculoskeletal fibromatoses: radiologic-pathologic correlation. Radiographics. 2009;29 (7): 2143-73.
- Soper JR, De Silva, M. Infantile myofibromatosis: a radiological review. Pediatric Radiol . 1993; 23: 189-194.
- Hatzidaki E , Korakaki E , Voloudak, A , Daskaloyannak, M , Manoura A, and Giannakopoulou C. Infantile Myofibromatosis with Visceral Involvement and Complete Spontaneous Regression. Jf Dermatol. 2001; 28: 379-382.
- Calvo-Garcia M, Foong-Yen L, Stanek J. Congenital peribronchial myofibroblastic tumor: prenatal imaging clues to differentiate from other fetal chest lesions. Pediatric Radiol. 2014; 44: 479-483.
- Naffa L, Khalifeh I, Salman R, et al. Infantile myofibromatosis: review of imaging findings and emphasis on correlation between MRI and histopathogical findings. Clin Imaging. 2018; 8(54):40-47.
- Lee JC, Thomas JM, Phillips S et al. Aggressive fibromatosis: MRI features with pathologic correlation. AJR Am J Roentgenol. 2006; 186 (1): 247-254.
Citation
A E, I G, RB T, AJ T, J NK. Infantile Myofibromatosis: Prenatal and Postnatal Imaging Features. Appl Radiol. 2021;(3):44-46.
May 4, 2021