Inflammatory Myofibroblastic Tumor
Images
CASE SUMMARY
A 2-year-old female presented to the emergency department with parental concerns regarding persistent, painless left upper-extremity weakness. The patient refused to lift her left arm above her head or to extend it in front of her body to pick up objects. Labs obtained included CBC with differential, CMP, ESR/CRP, uric acid, and LDH. All were unremarkable with the exception of moderate neutropenia (ANC 300) and mild elevation in LDH (255). Urine HVA and VMA were within normal limits.
IMAGING FINDINGS
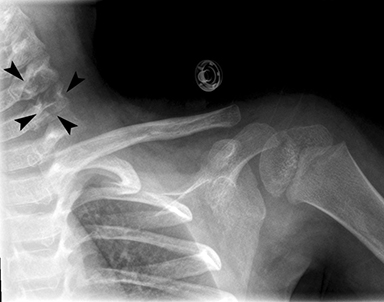
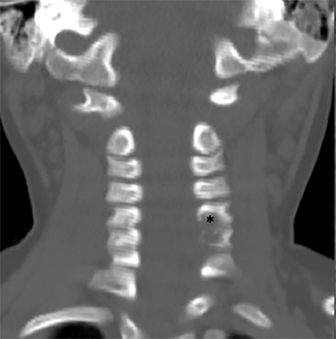
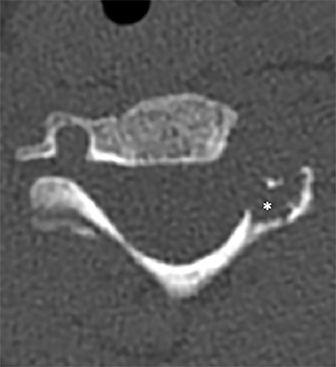
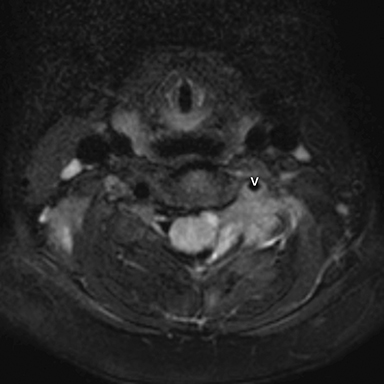
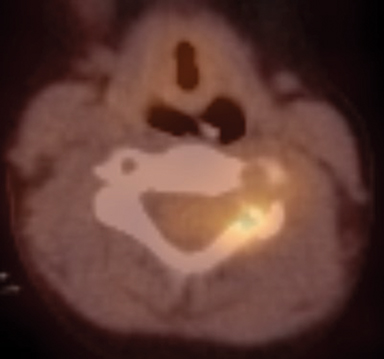
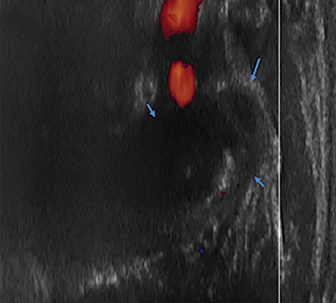
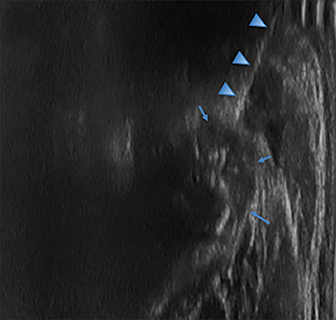
Radiographs of the left clavicle showed lytic lesions of the left C5 and C6 transverse processes (Figure 1). Subsequent cervical MRI and CT studies were performed, helping to further characterize the lesion as a soft-tissue cervical spinal mass. Coronal and axial CT images of the cervical spine without contrast showed bone destruction of the left C5 and C6 transverse process and lamina with widening of the left C5/C6 neural foramen extending into the left transverse foramen (Figure 2). Axial T1 postcontrast MRI of the cervical spine established the presence of abnormal soft-tissue contrast enhancement centered within the left C5/C6 neural foramen with extension into the extra-medullary space within the spinal canal and approximately 270 degrees of encasement around the left vertebral artery (Figure 3). Transverse PET/CT imaging (Figure 4) showed abnormal FDG uptake within the mass with an SUV of 4.3. Color Doppler ultrasound was performed to locate the left vertebral artery and plan the biopsy route (Figure 5) with subsequent needle biopsy of the mass using ultrasound guidance via an anterolateral approach (Figure 5).
DIAGNOSIS
Inflammatory myofibroblastic tumor (IMT).
Differential diagnosis of the cervical lesion includes nodular fasciitis, myofibroma, fibrodysplasia ossificans, inflammatory liposarcoma, spindle cell caricinoma, osteomyelitis, and neuroblastoma.
DISCUSSION
Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of mesenchymal origin most commonly affecting children and young adults. Histologically, IMT is made up of smooth muscle spindle cells embedded in a myxoid or fibrous stroma. Histologically there are inflammatory cells such as plasma cells, lymphocytes, or eosinophils; the neoplasm was initially categorized under a broad classification termed “inflammatory pseudotumors,” which includes many different reactive or infectious entities. 1 The gross appearance of IMT is described as soft and rubbery with a white or gray cut-surface.
This tumor is uncommon, with 150-200 reported pediatric cases in the United States.1 IMT is most commonly found in the lungs or abdomen, but it has been reported in other locations, including two recent reports of occurrence in the head and neck region.2 A review of 84 cases by Coffin et al (1995) demonstrated that extra-pulmonary involvement presents at a younger age than does pulmonary involvement.3 In approximately 30% of patients, diagnosis is preceded by a syndrome of intermittent fever, malaise, weight loss, and night sweats, along with laboratory findings of hypo- or normochromic anemia and elevated ESR. These symptoms are thought to be caused by an elevation of the cytokine IL-6, leading to a systemic inflammatory response.1
Immunohistochemical characteristics of IMT are variable and nonspecific, but can include positivity for smooth muscle actin (SMA) (80-90% of cases) and cytokeratins AE1, AE3, and CAM 5.2 (33% of cases).4 About one-half of all IMTs contain an anaplastic lymphoma kinase (ALK) gene rearrangement on chromosome 2 at band 2p23, associated with numerous gene partners.4 Other gene mutations infrequently seen in IMT include p53 and MDM2.5 A small portion of ALK-negative IMTs show rearrangement in the ROS1 gene (up to 10%). 4
Traditionally, subclassification of myofibroblastic tumors has been notoriously difficult due to a lack of specific immunohistochemical markers. The most common immunohistochemical findings are a strong positivity for smooth muscle actin and desmin, and negativity for h-caldesmon.6 Differential diagnosis includes nodular fasciitis, myofibroma, fibrodysplasia ossificans, fibrous histiocytoma, and spindle cell carcinoma. Given the variable and unpredictable histologic characterization of IMTs, the correct diagnosis is made on the basis of molecular and cytogenetic analysis using techniques such as FISH and PCR.
On CT imaging, IMT can present as a soft-tissue mass slightly hypodense to surrounding muscle, although variations in attenuation are often seen. The presence of adjacent fat stranding is common and likely due to an associated inflammatory process.7 Calcifications and/or central necrosis are rare of this type of tumor, suggesting a more benign course.
MR imaging tends to reveal intermediate-to-low signal intensity on both T1- and T2-weighted images as a result of the associated fibrosis. Depending on the specific anatomical location, lesions can be either well-circumscribed or ill-defined and infiltrating. Lesions in the head and neck region can appear aggressive, resulting in ill-defined bony erosions. Imaging can reveal expansile osteolytic lesions with an adjacent soft-tissue component. Contrast enhancement patterns are usually nonspecific and variable.8
IMT is classified by the World Health Organization as having ‘intermediate biological potential’ mostly due to a tendency for local recurrence. 9 Multiple sources have reported a recurrence rate of 25% for extra-pulmonary lesions.1,2 The mainstay of treatment is surgical resection; however, this can prove difficult depending on location and extension of the lesion. For unresectable lesions, the use of targeted molecular therapy (ie, crizotinib, tyrosine kinase inhibitor) has been utilized and proven efficacious.8 Narayanappa et al (2018) reported 2 patients free of disease at 3 years’ follow-up after complete resection.
CONCLUSION
Inflammatory myofibroblastic tumors are rare neoplasms of mesenchymal origin primarily seen in children and young adults. Diagnosis can be challenging due to nonspecific clinical presentation and variable anatomic locations of tumors. Diagnosis is primarily one of exclusion and confirmed by biopsy; however, ALK gene rearrangements are seen in up to 50% of cases. Imaging findings are nonspecific as well, and include variable attenuation values on CT and intermediate-to-low signal intensity on MRI. Lesions in the head and neck can appear malignant, with erosion into bone and infiltration into adjacent soft tissue. Treatment is based on complete surgical excision and the use of specific molecular targets for unresectable lesions. This case illustrates the importance of evaluating all of the images within a study. The lytic lesions revealed on X-ray of the clavicle was initially missed.
REFERENCES
- McDermott, M. Inflammatory myofibroblastic tumour. Seminars in Diag. Path. 2016; 33(6): 358-366. https://www.clinicalkey.com/ - !/content/journal/1-s2.0-S0740257016300661?scrollTo=%23hl0000255. Accessed July 9, 2018.
- Narayanappa H, Low T, Calderon ACV, Selinger C, Gupta R. Inflammatory myofibroblastic tumours of the head and neck. Pathology. 2018; 50(3): 356-358. https://www.clinicalkey.com/ - !/content/journal/1-s2.0-S0031302517303410. Accessed July 9, 2018.
- Coffin CM, Watterson J, Priest JR, Dehner LP. Extra Pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am. J Surg. Pathol. 1995;19(8):859-872.
- Doyle LA, Hornick JL. Immunohistochemistry of Neoplasms of Soft Tissue and Bone. In: Diagnostic Immunohistochemistry. 5th ed. Elsevier; 2019. https://www.clinicalkey.com/ - !/browse/book/3-s2.0-C20150054016. Accessed July 9, 2018.
- Jain S, Zhang DY, Xu R, Pincus MR, Lee P. Molecular Genetic Pathology of Sarcoma. In: Henry’s Clinical Diagnosis and Management by Laboratory Methods. 23rd ed. Elsevier; 2017. https://www-r2library-com.mwu.idm.oclc.org/Resource/Title/0323295681. Accessed July 9, 2018.
- Goldblum JR. Soft Tissues. In: Rosai and Ackerman’s Surgical Pathology. 11th ed. Elsevier; 2018. https://www.clinicalkey.com/-!/content/book/3-s2.0-B978032326339900041X?scrollTo=%23hl0002764. Accessed July 9, 2018.
- Hourani R, Taslakian B, Shabb NS, Nassar L, Hourani MH, Moukarbel R, Sabri A, Rizk T. Fibroblastic and Myofibroblastic tumors of the head and neck: Comprehensive imaging-based review with pathologic correlation. European Journal of Radiology. 2015; 84(2): 250-260. https://www.clinicalkey.com/ - !/content/journal/1-s2.0-S0720048X14004823?scrollTo=%23hl0000515. Accessed July 9, 2018.
- Surabhi VR, Chua S, Patel RP, Takahashi N, Lalwani N, Prasad SR. Inflammatory Myofibroblastic Tumors. Radiologic Clinics of North America. 2016; 54(3): 553-563. https://www.clinicalkey.com/ - !/content/journal/1-s2.0-S0033838915002390?scrollTo=%23hl0000486. Accessed July 9, 2018.
- Coffin CM, Fletcher JA. Inflammatory myofibroblastic tumor. In: Retcher CDM, Unni KK, Mertens F, eds. World Health Organization classification of tumors: Pathology and genetics of tumors of soft tissue and bone. Lyon: IARC Press, 2002: 91-93.
Citation
M G, D A, CS S, R K, SA J, T A, A T, R T.Inflammatory Myofibroblastic Tumor. Appl Radiol. 2020; (1):56D-56F.
January 23, 2020