Multimodality Evaluation of Fetal Congenital Diaphragmatic Hernia and Its Mimics
Images

SA-CME credits are available for this article here.
Congenital diaphragmatic hernia (CDH) is a rare fetal chest abnormality encountered on obstetric ultrasound often with major associated postnatal consequences. CDH usually presents as a chest mass that may be difficult to distinguish from other fetal chest masses. Other chest masses that may be confused with CDH include congenital pulmonary airway malformation (CPAM), and bronchopulmonary sequestration (BPS). These entities will be briefly discussed.
The focus of this article will be CDH and the radiologist’s evolving role in imaging of this entity. We will discuss salient ultrasound and MR imaging findings that enable a diagnosis of fetal CDH and describe parameters used to predict newborn survival and prognosis.
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia is a chest abnormality that is associated with high fetal mortality. The incidence of CDH is approximately 2-4 in 10,000 live births and 40-50% are associated with other congenital abnormalities.1 CDH always causes some degree of pulmonary hypoplasia due to mass effect within the thorax.2 The prognosis of CDH is dependent on other associated abnormalities and the severity (size) of the CDH. “Liver up” is an important but independent risk factor. However, mortality in the newborn is typically from pulmonary hypertension.
The ability of the diagnostic imager to estimate the size and contents of the CDH and, more importantly, to estimate the amount of residual lung by either ultrasound or MRI is of paramount importance in planning therapy.
Therapy can include newer techniques such as fetal endoscopic tracheal occlusion (FETO), which will be briefly discussed later. For over 20 years the mortality of CDH had plateaued at approximately 30%. However, in the past 5 years the mortality has dropped to 25.8%.3
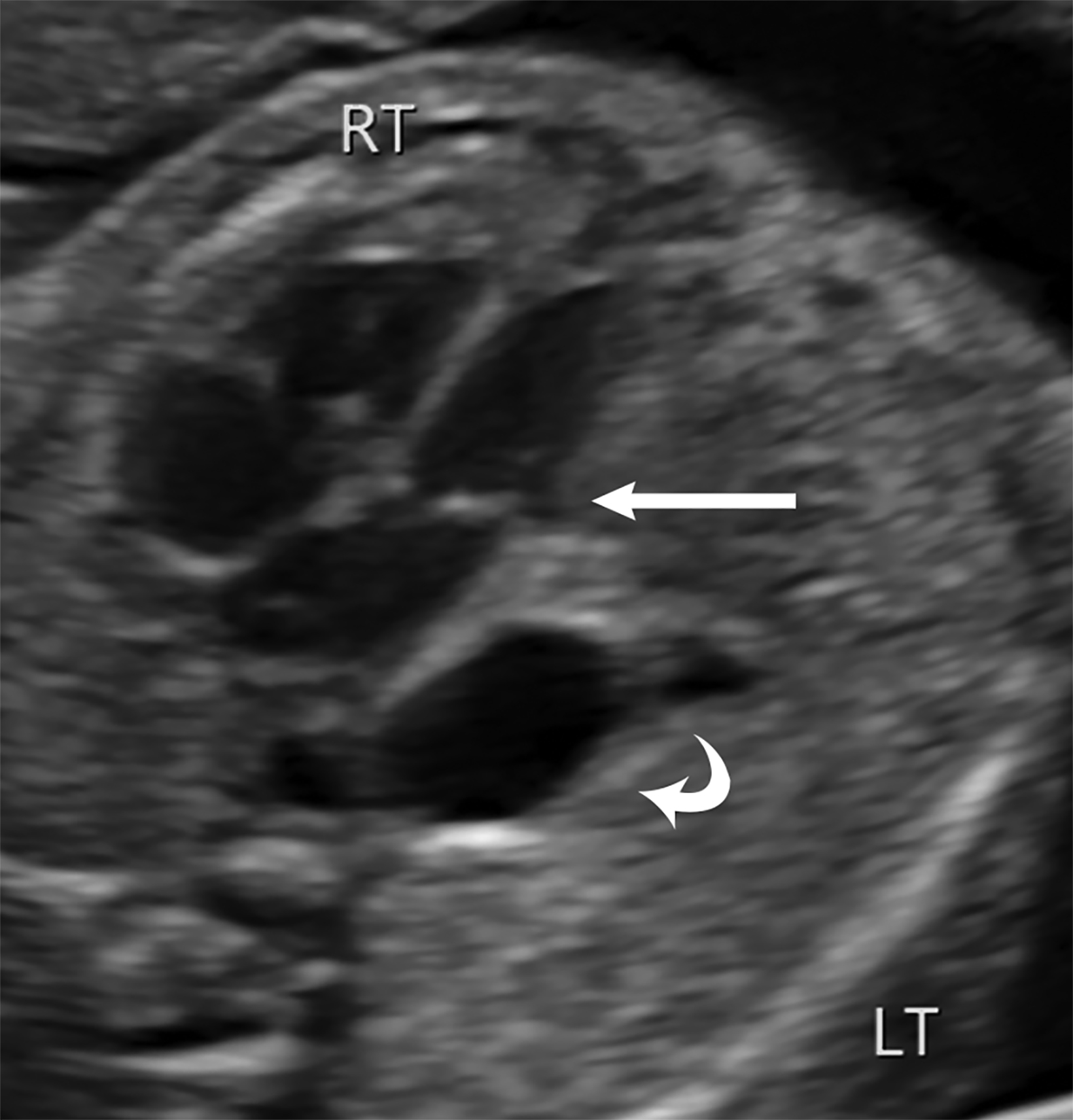
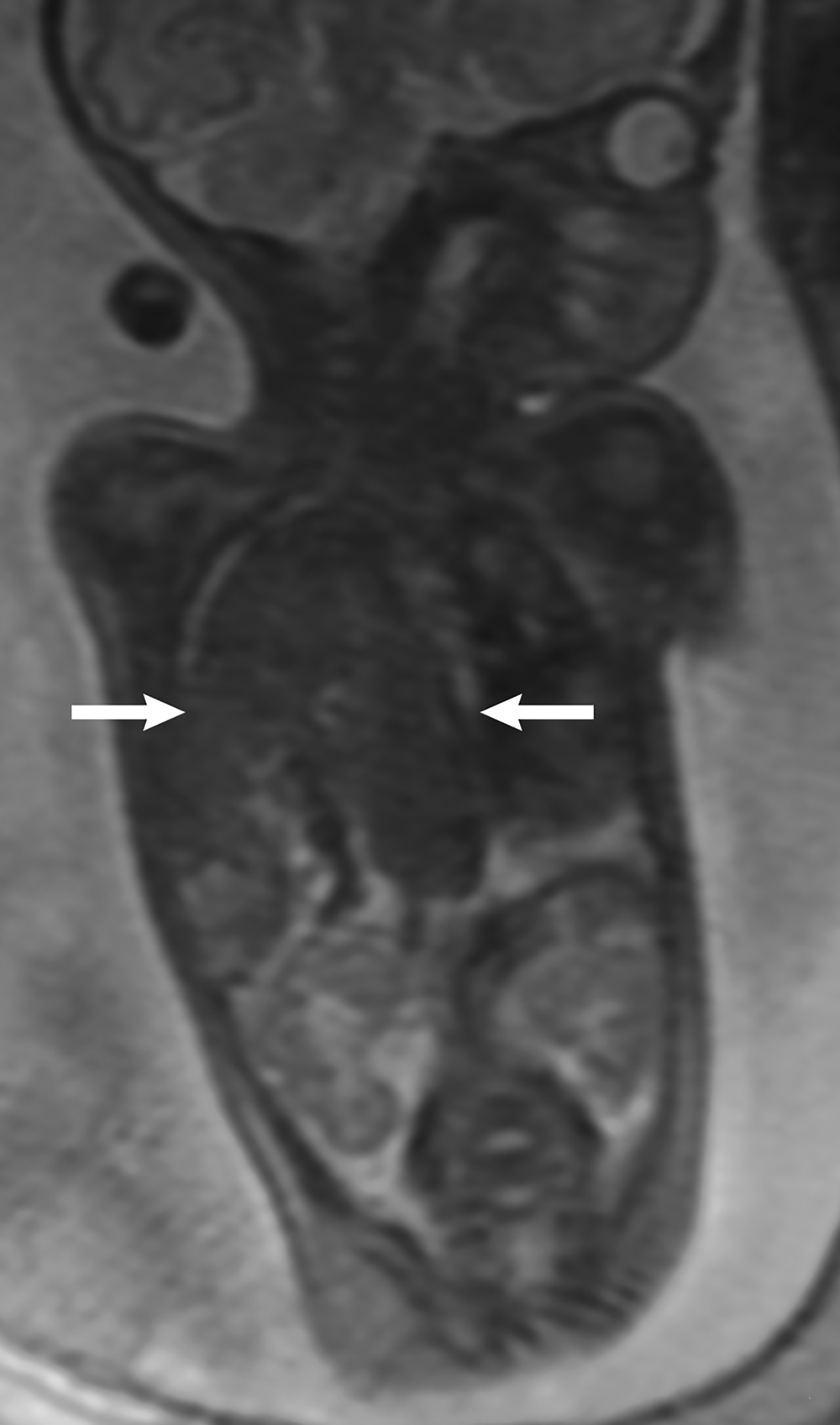
Most CDH cases are suspected on routine fetal anatomy ultrasound, with a reported mean age at diagnosis of 22 – 24 weeks.2 Ultrasound is the initial imaging modality for diagnosis and evaluation of CDH. Multiple sonographic features have been used to diagnose fetal CDH. These include direct signs such as herniated bowel loops, stomach, and/or liver displaced into the thoracic cavity.
Indirect signs include abnormal position of the heart with mediastinal shift secondary to mass effect.4 Two widely used ultrasound parameters are utilized to predict newborn survival and the need for postnatal extracorporeal membrane oxygenation (ECMO), including the lung area to head circumference ratio (LHR) and the location of the fetal liver.4
Fetal MR imaging is becoming more widely used and it is important that the radiologist knows some of the important findings and prognostic features of fetal MRI. Fetal MR can corroborate the ultrasound findings with increased diagnostic accuracy and provide added prognostic value with risk assessment not possible on ultrasound.5
The utility of MRI also includes improved anatomic assessment, determination of specific hernia type, and evaluation and exclusion of associated malformations and complications. Calculation of lung volumes and liver herniation measurements performed on MRI are key to prenatal counseling and perinatal planning.5
Differential for Fetal Chest Masses
The two most common fetal chest masses that may mimic congenital diaphragmatic hernia include CPAM and BPS.6 Less common etiologies of a fetal chest mass include congenital high airway obstruction syndrome (CHAOS), congenital bronchogenic cyst, bronchial atresia, pulmonary arteriovenous malformation, congenital pulmonary lymphangiectasia, pulmonary hypoplasia-aplasia, mediastinal teratoma, mediastinal lymphatic malformation, and congenital hydrothorax.6 We will focus our discussion on the three most common fetal chest masses that may have similar but distinguishing features from CDH. A few of the other abnormalities will also be presented.
Congenital Pulmonary Airway Malformation
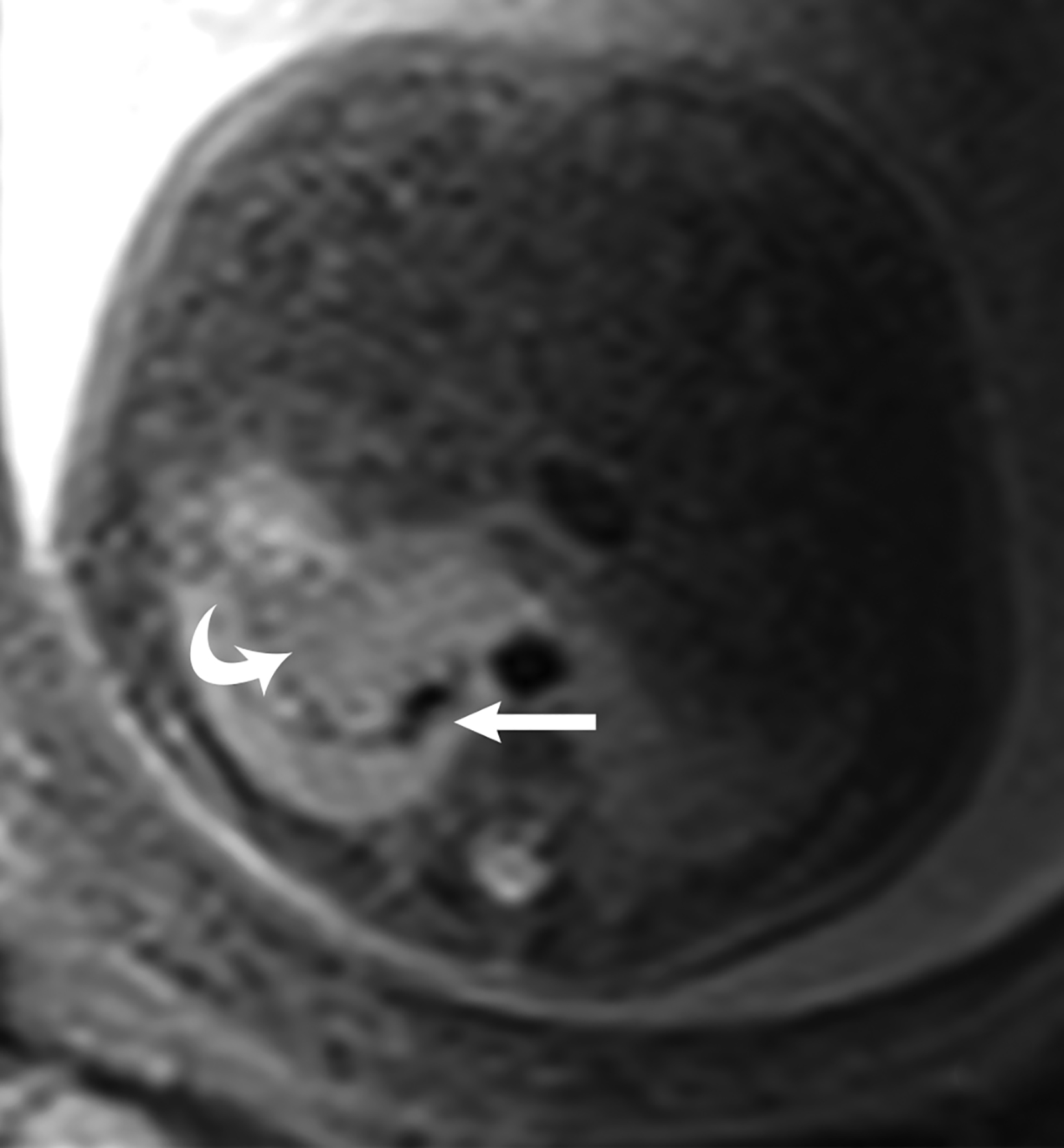
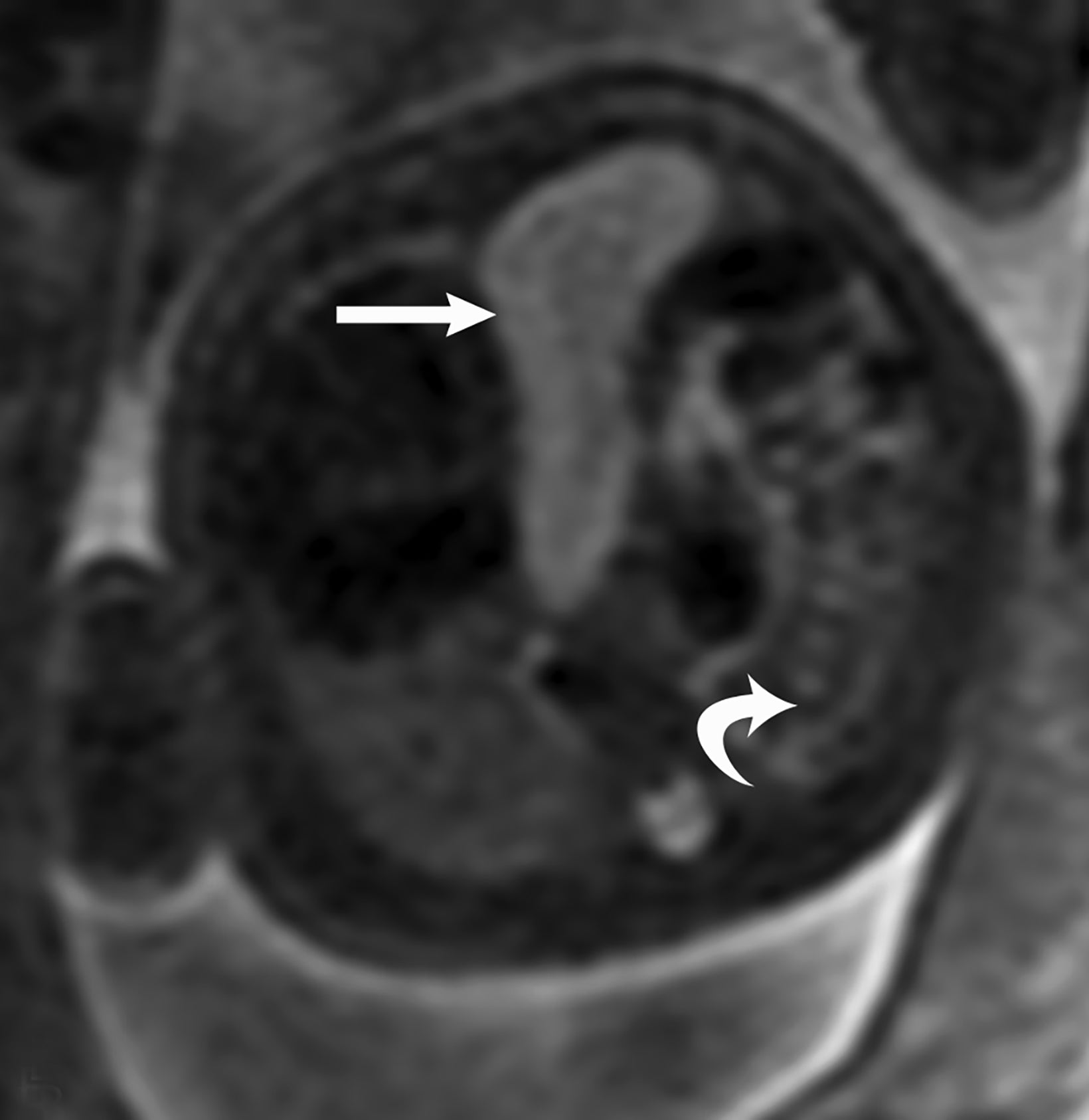
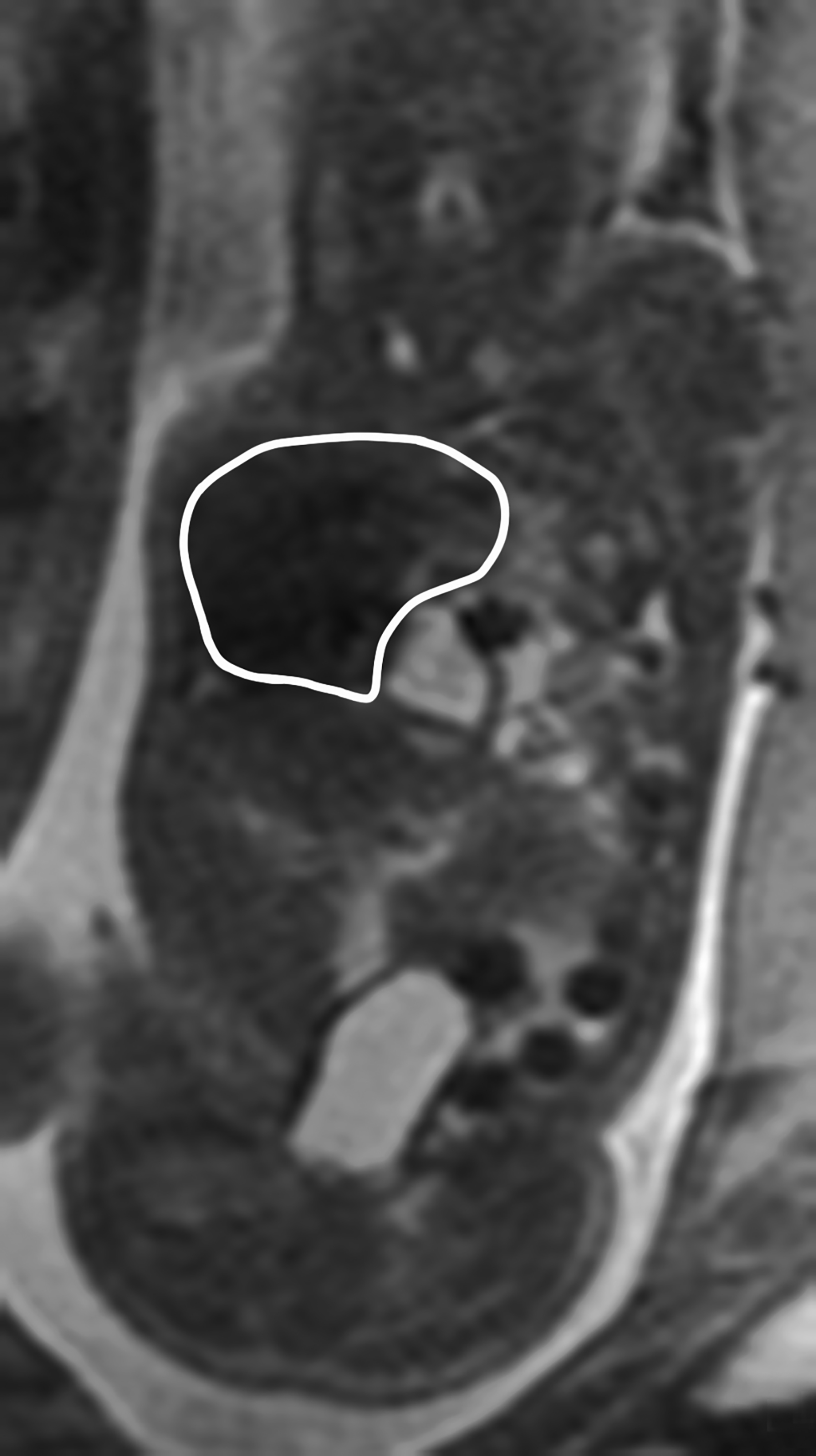
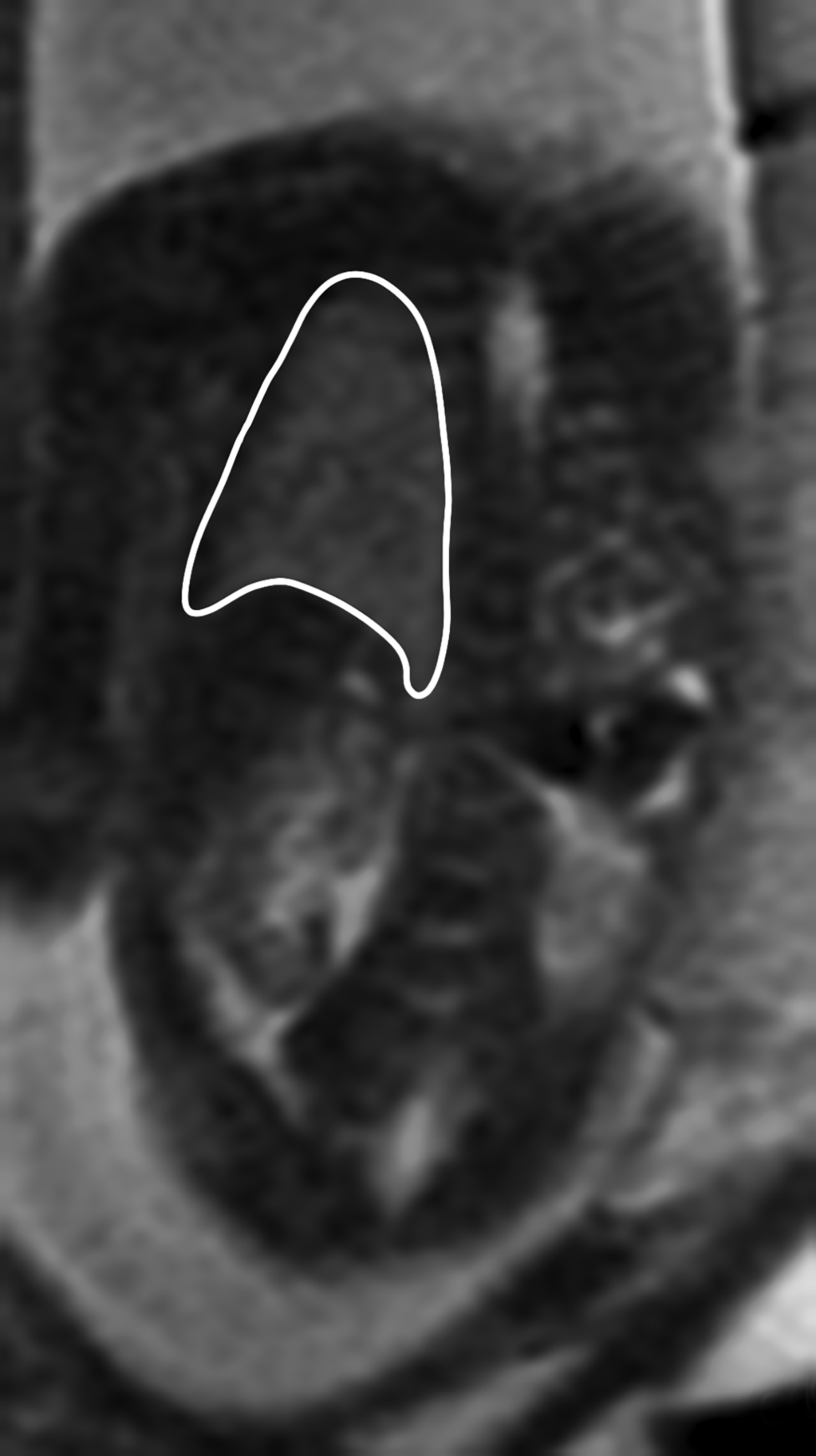
Congenital pulmonary airway malformation, previously known as congenital cystic adenomatoid malformation, is a congenital anomaly of the lower respiratory tract.6,7 CPAM is a mass of lung tissue with varying degrees of cystic change that may communicate with the tracheobronchial tree and has normal pulmonary arterial supply and venous drainage (Figures 1, 2).
There are 3 original histopathologic types of CPAM, classified based on size and number of cysts based upon the Stocker classification.7 More recently this classification system has been expanded to 5 subtypes.8 These are classified as Type 0 through IV. Type I is the most common occurring in 60%-65% of cases and has multiple large cysts or a single dominant cyst.8 All CPAMs have variable size cysts. Imaging findings vary depending on the subtype. CPAM lesions are typically unilateral and more commonly found in the lung bases.
The majority of CPAM spontaneously decrease in size and occasionally resolve in the third trimester. Many infants who have a prenatal diagnosis of CPAM are asymptomatic at birth. However, some CPAMs may exert mass effect on the lungs and mediastinal structures. There may be pulmonary hypoplasia if the CPAM is large. Fetal hydrops may occur with CPAM which has been shown to be a predictor of poor fetal outcome.7
Bronchopulmonary Sequestration
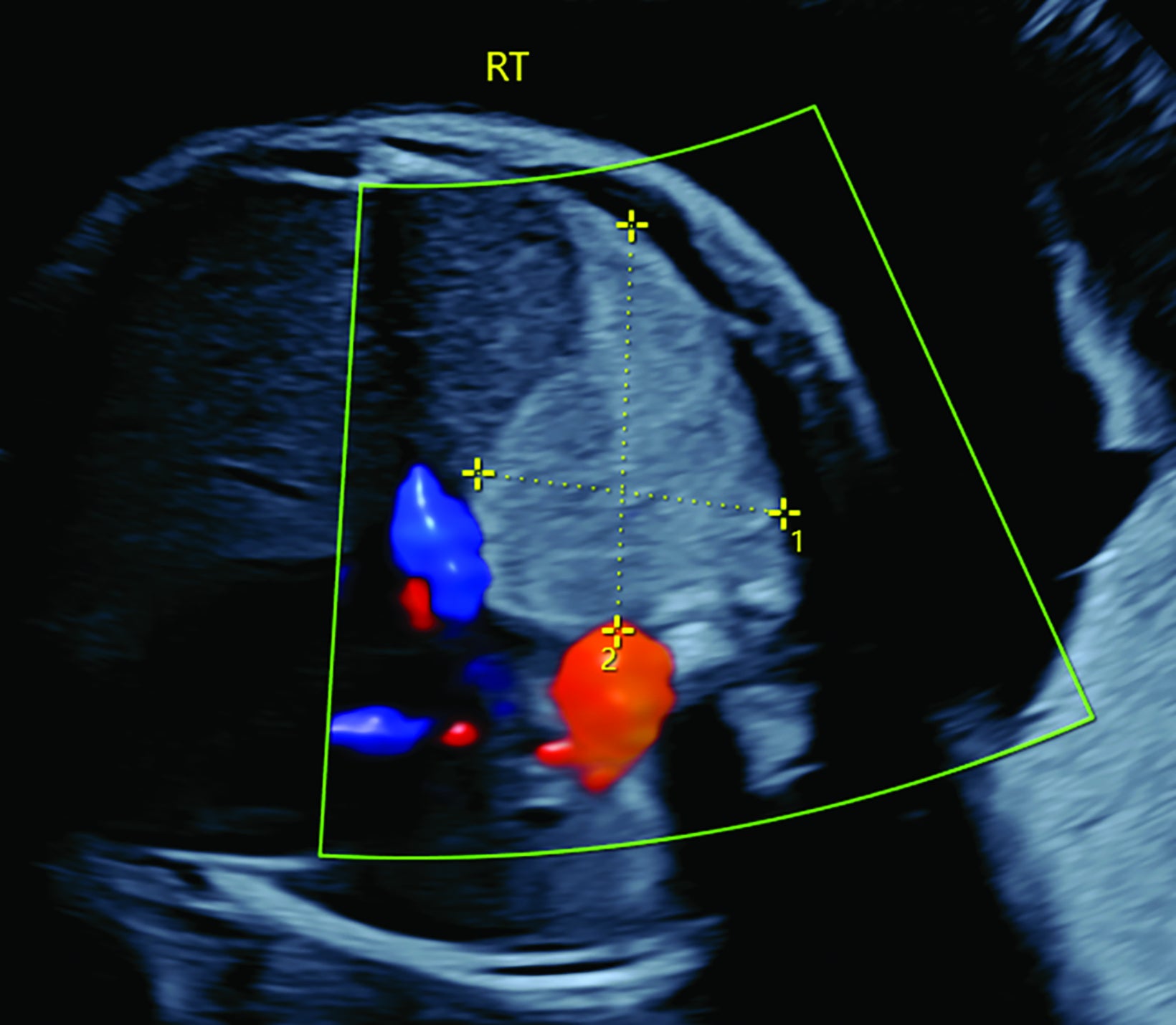
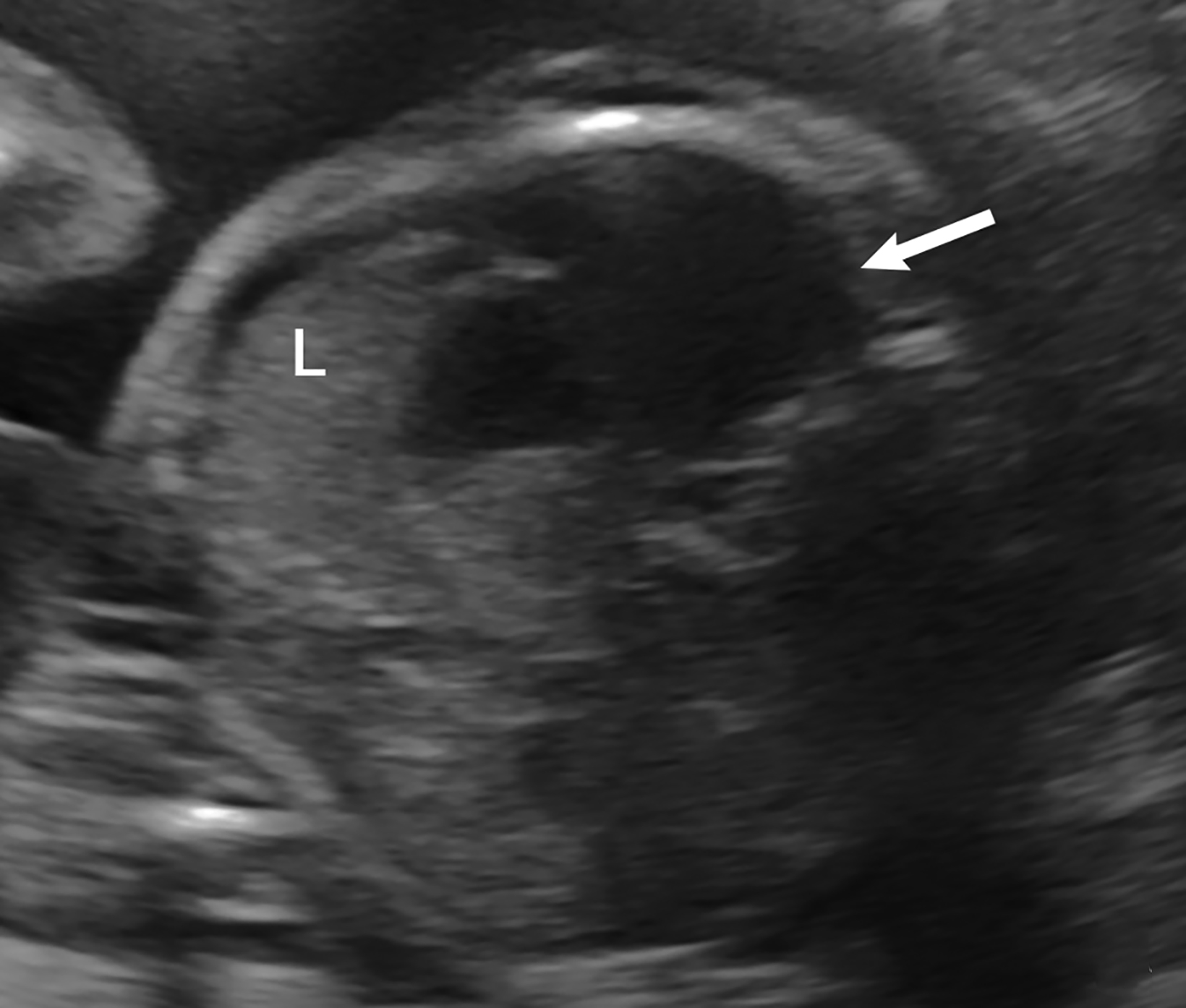
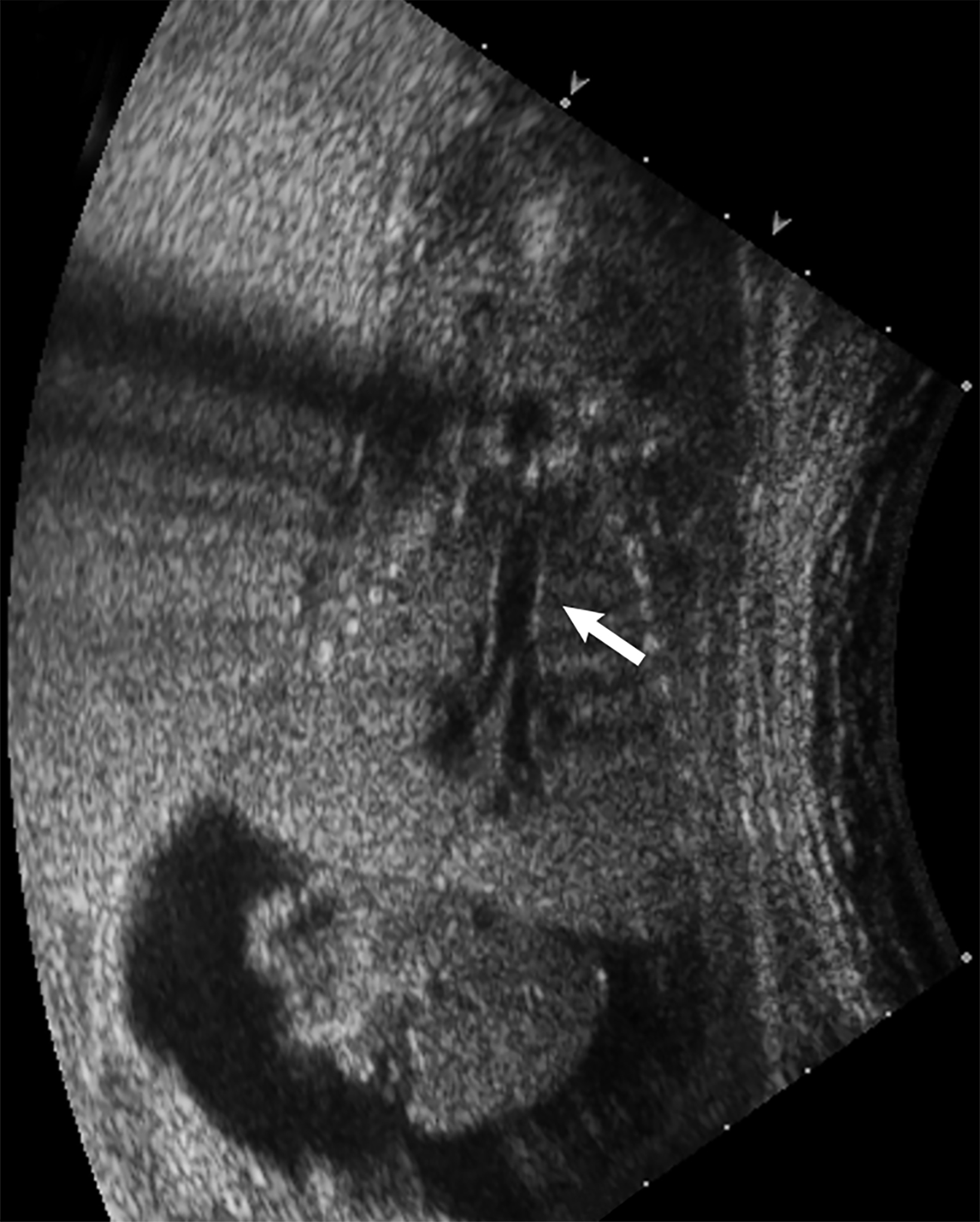
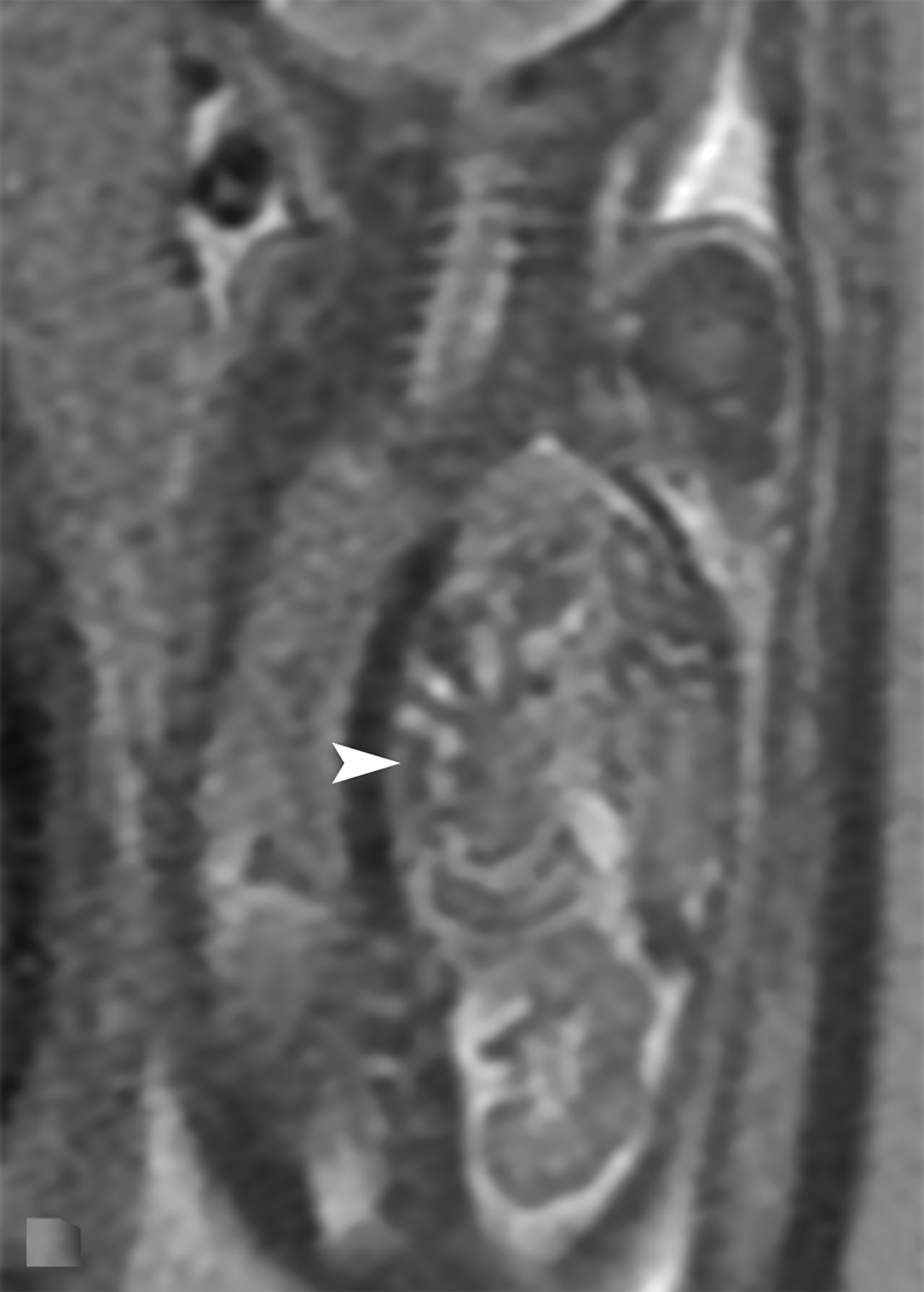
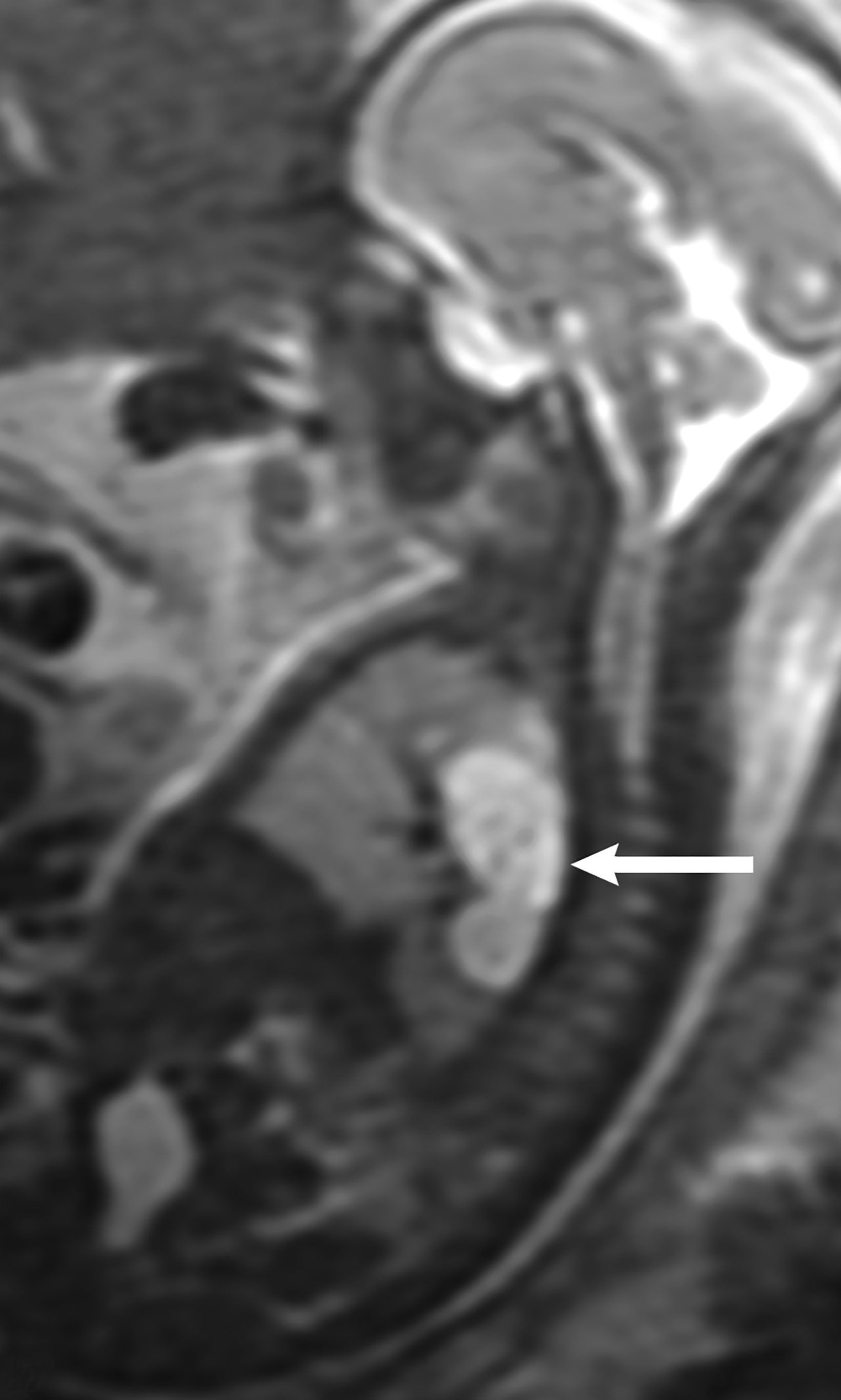
Bronchopulmonary sequestration is characterized by abnormal bronchopulmonary tissue that does not communicate with the tracheobronchial tree and is not supplied by pulmonary vasculature.6,7 Bronchopulmonary sequestration is within the differential diagnosis for CPAM or CDH. Unlike CPAM, there is lack of communication with the tracheobronchial tree and lack of vascular supply from the pulmonary artery. Sequestration is usually fed via a systemic artery.7 Hybrid lesions are masses that contain features of both CPAM and bronchopulmonary sequestration. Key imaging findings of BPS include a well-defined solid homogenous mass with systemic vascular supply.7 The feeding artery is usually better visualized on US compared to MRI (Figure 3). On MRI, the mass will demonstrate T2 signal higher in comparison to the normal lung.7 (Figure 4).
Of the two subtypes, intralobar BPS in more common (75% of cases) and has its own visceral pleura. Extralobar sequestration has no viscera pleura encasing the abnormal tissue.7 This sub-type is more commonly identified in utero or the newborn, while intralobar BPS can be identified in utero it often presents in childhood. BPS is more frequently found in the lower lobes, most commonly the left lower lobe.7A fetal pleural effusion may be secondary to BPS.
Development of serious complications like hydrops, secondary to BPS, may prompt early delivery. Postnatal surgical excision is recommended for infants due to the risk of infection development or malignant transformation.6 The key discriminating features of the three most common fetal chest masses (CDH, CPAM, BPS) are summarized in Table 1.
Congenital Hydrothorax
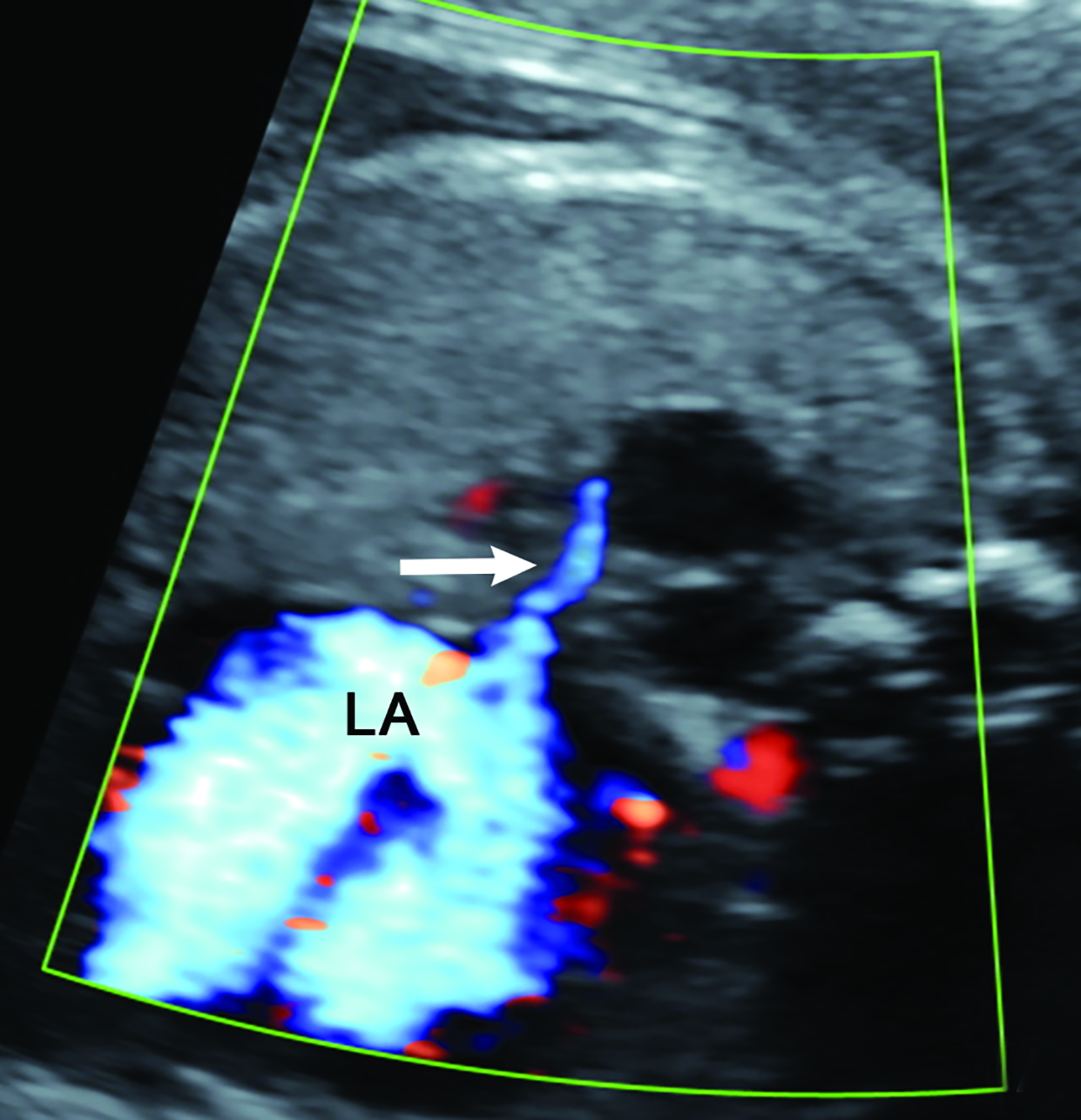
Congenital hydrothorax is defined as pleural fluid within the fetal thorax (Figure 5). Fetal pleural effusion may be unilateral or bilateral.7On ultrasound, the hydrothorax will occupy either one or both thoracic cavities. It appears as an anechoic region that surrounds other thoracic structures such as the lung or heart. Primary congenital hydrothorax is rare and indicates fetal pleural effusion without any underlying abnormality. This may result from a chylous leak due to abnormal lymphatic ducts; thus, the pleural fluid contains chyle. Congenital hydrothorax may be secondary to a fetal chest mass or other abnormalities in approximately 40% of cases.
These include a wide spectrum of abnormalities such as CDH, CPAM, BPS, Turner syndrome, Down syndrome, cystic hygroma, and cardiac anomalies.7 It is important to recognize that an underlying mass such as sequestration can present as a hydrothorax.
Mass effect from the fluid collection may result in pulmonary hypoplasia.
On MRI, T1 and T2 signal intensity of the fluid can vary depending on fluid content but will typically be hypointense on T1 images and hyperintense on T2 images.7 Careful inspection of the MR for an underlying etiology is important.
Congenital High Airway Obstruction
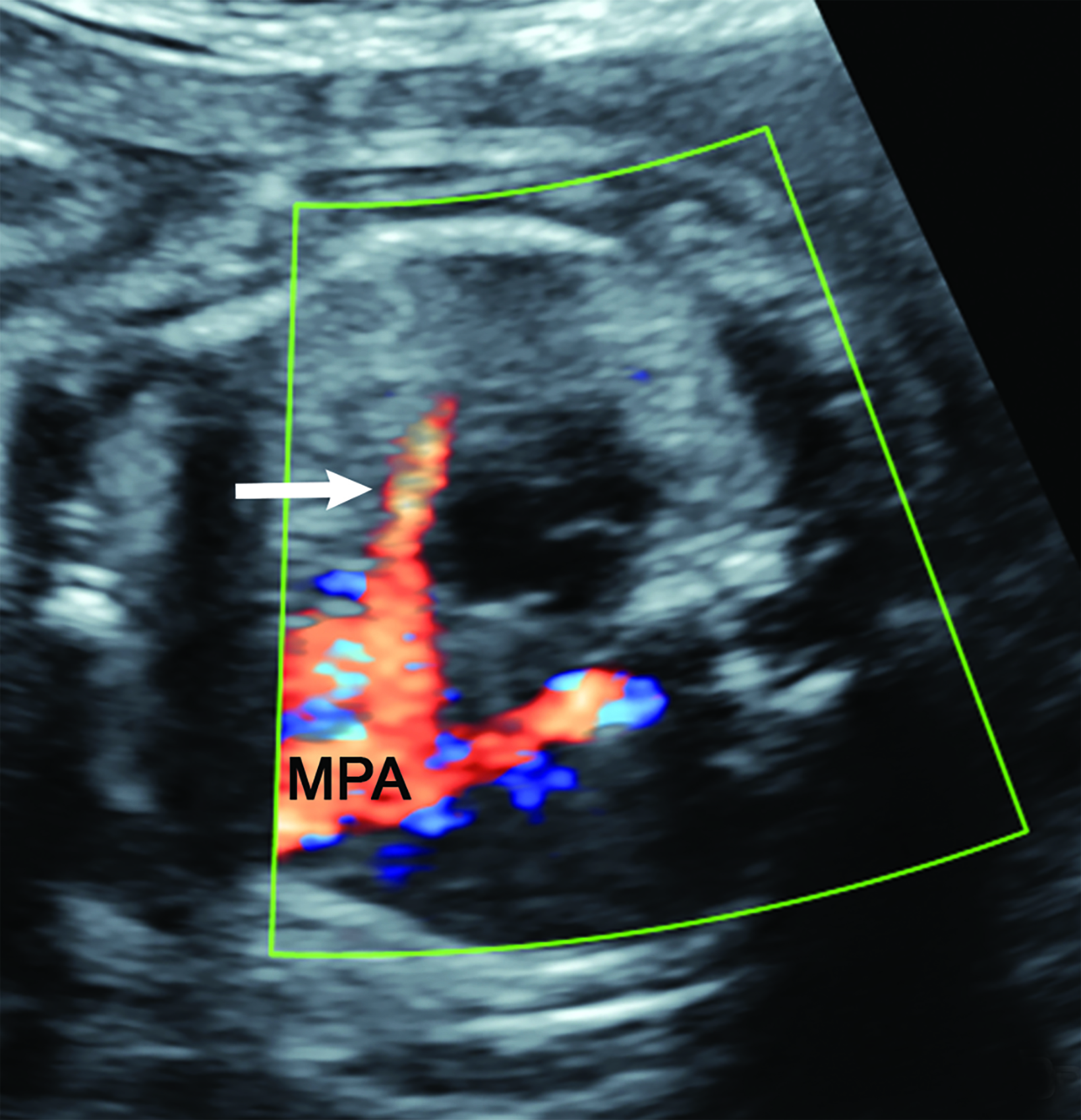
Congenital high airway obstruction syndrome is defined as obstruction of the upper airway, either the larynx or trachea, and may be caused by atresia, stenosis, or a web.7 On obstetric ultrasound, lungs are symmetrically enlarged and echogenic, resulting in a flattened or everted diaphragm.7 (Figure 6). The heart is typically small and midline. There may be ascites, which can be massive.
CHAOS is fatal unless prompt intervention is performed at the time of delivery. The ex utero intrapartum treatment (EXIT) procedure is used to preserve uteroplacental circulation as cardiopulmonary bypass for the fetus by the placenta until an airway can be surgically created.
Congenital Diaphragmatic Hernia Diagnostic and Prognostic Features
Ultrasound — Diagnostic Features
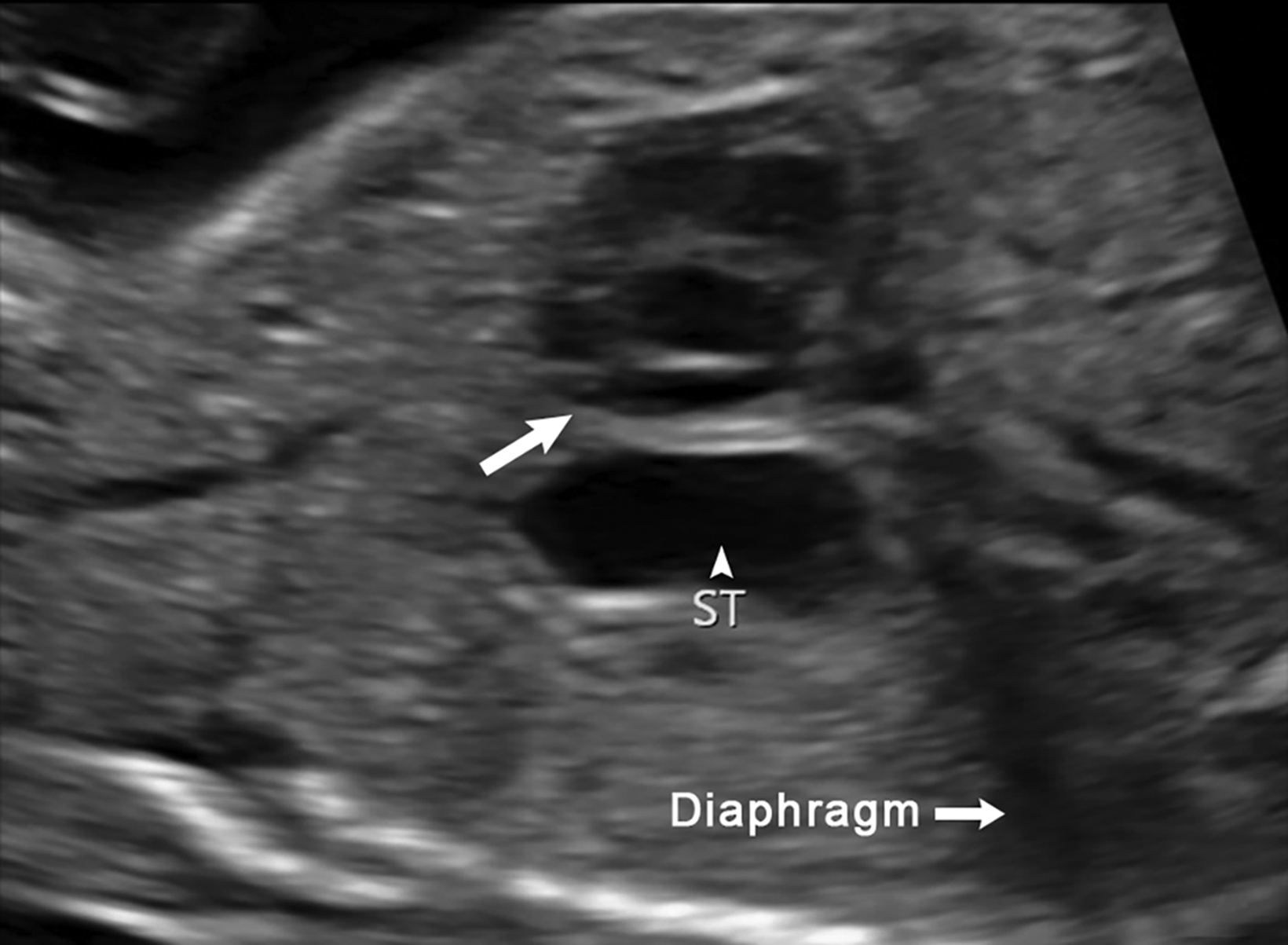
Ultrasound is the first line imaging modality for evaluation of CDH. Diagnostic ultrasound features include the presence of a chest mass, loss of the diaphragmatic boundary on diaphragm view, and abdominal contents in the chest.1,4 Multiple sonographic features have been used to diagnose fetal CDH. Some features are pathognomonic for CDH include: the presence of small bowel with peristalsis, stomach, and/or liver displaced in the chest. Secondary findings include deviation of the heart or mediastinum due to mass effect.4 (Figure 7).
Ultrasound — Prognostic Features
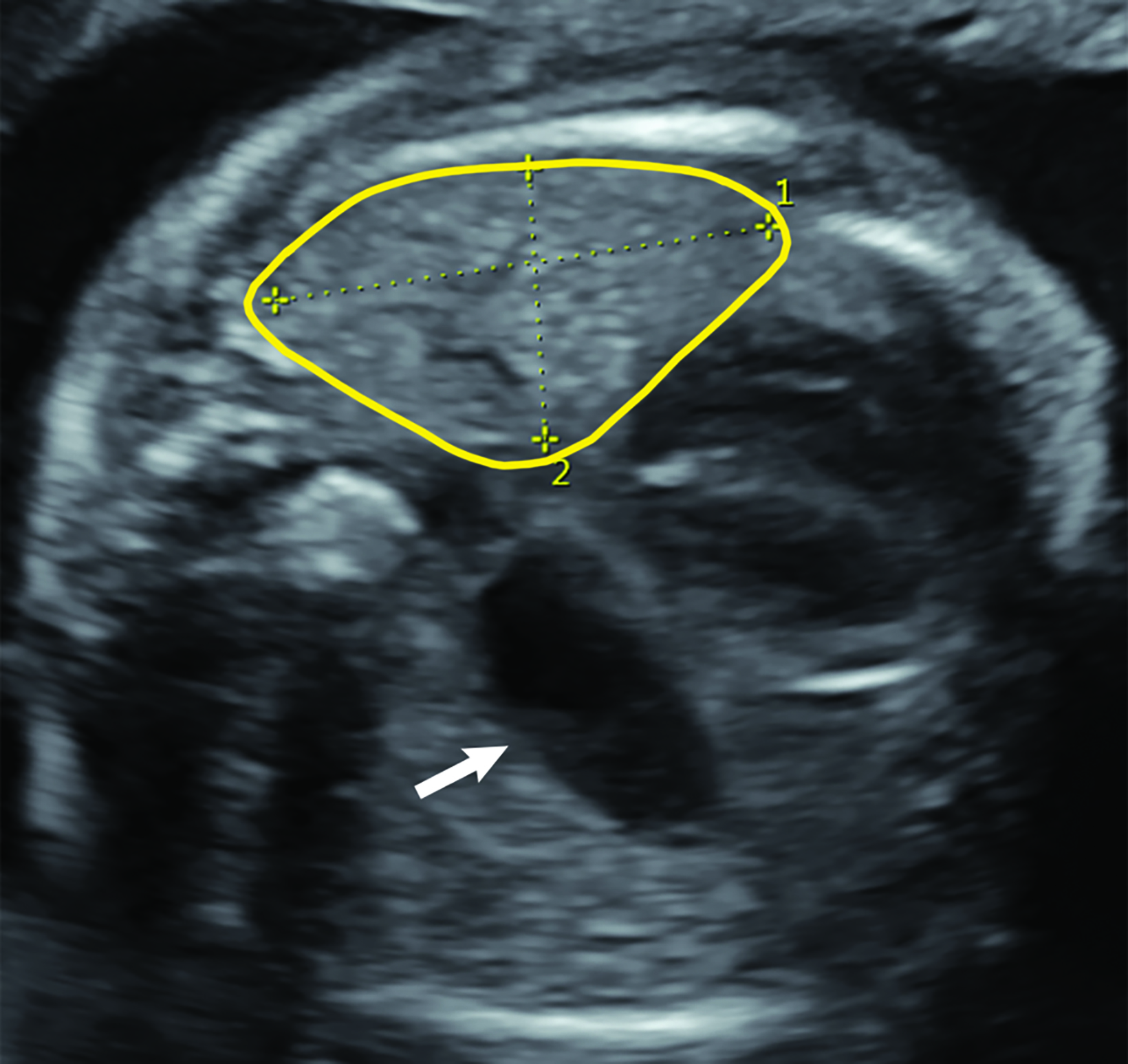
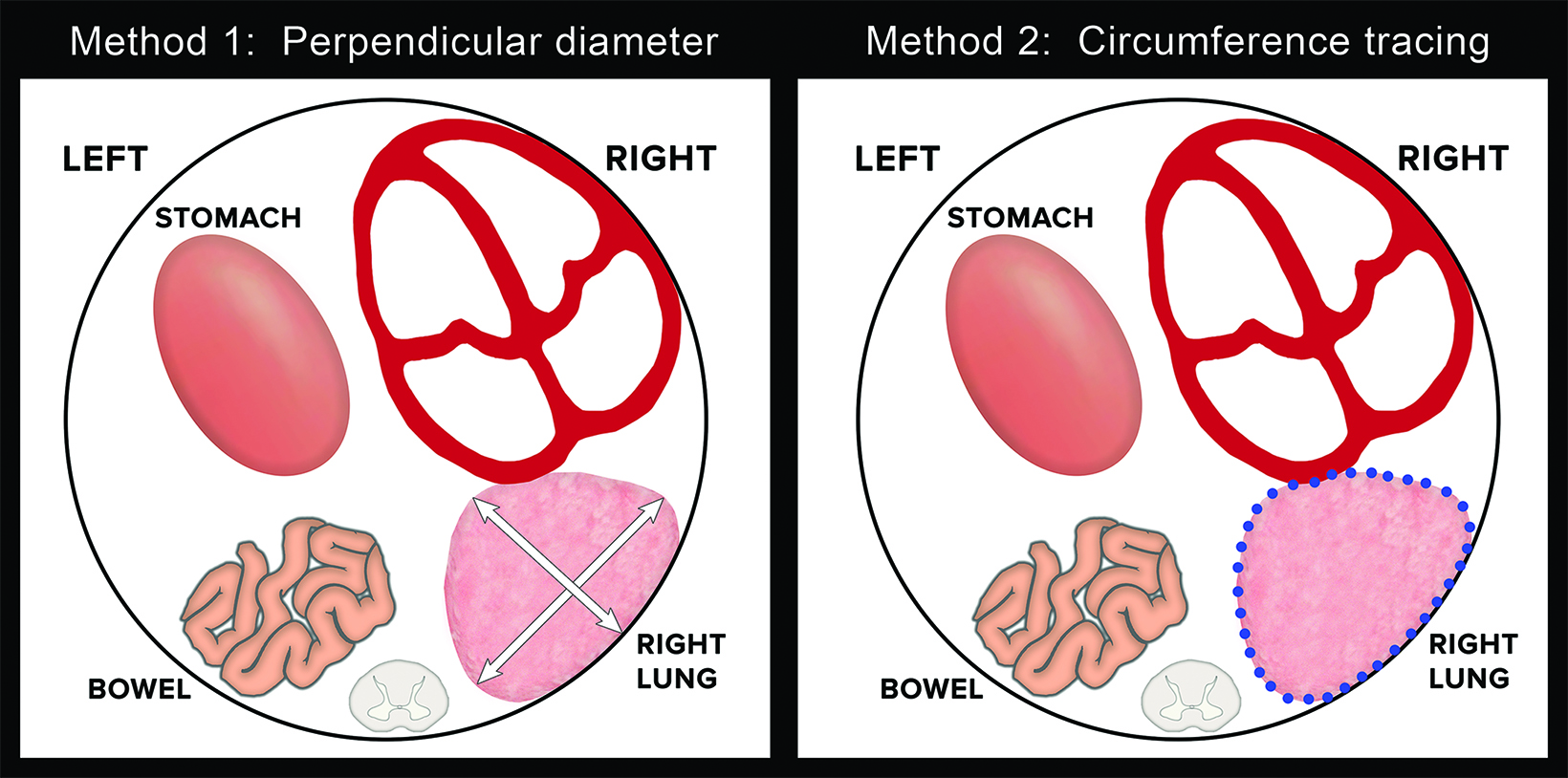
Ultrasound can also be used for prognostic assessment. The LHR is a sonographic measurement that is helpful to determine the residual lung and is a prognostic factor in determining fetal survival.4 (Figure 8) The cross-sectional area of the fetal lung is measured from a 4-chamber view of the heart on transverse scan.9 The cross-sectional lung area can be measured by the AP diameter method, the longest axis method, or the tracing method. Of these methods the anteroposterior method is considered least reliable of the methods. One calculator can be found at https://totaltrial.eu/?id=6. The cross-sectional area method, defined as the perpendicular diameter of the lung (Method 1), or by circumferential tracing of the lung border (Method 2) is depicted in Figure 9.7 The circumferential tracing method is a more reproducible method than methods. The lung area is then divided by head circumference to calculate the LHR. The LHR can then be compared to reference values. Another calculator can be found at perinatology.com (https://www.perinatology.com/calculators/LHR.htm).9 Based on different studies, there is 100% fetal mortality with LHR< 0.6-1 and 100% survival with LHR> 1.35-1.4.4
Other prognostic features that can be assessed on ultrasound include the location of the fetal liver. If the fetal liver is above the diaphragm, this can be associated with a worse prognosis, including lower rates of fetal survival and increased ECMO requirement. However, pulmonary hypertension is often the etiology of infant death.3
Magnetic Resonance Imaging — Diagnostic Features
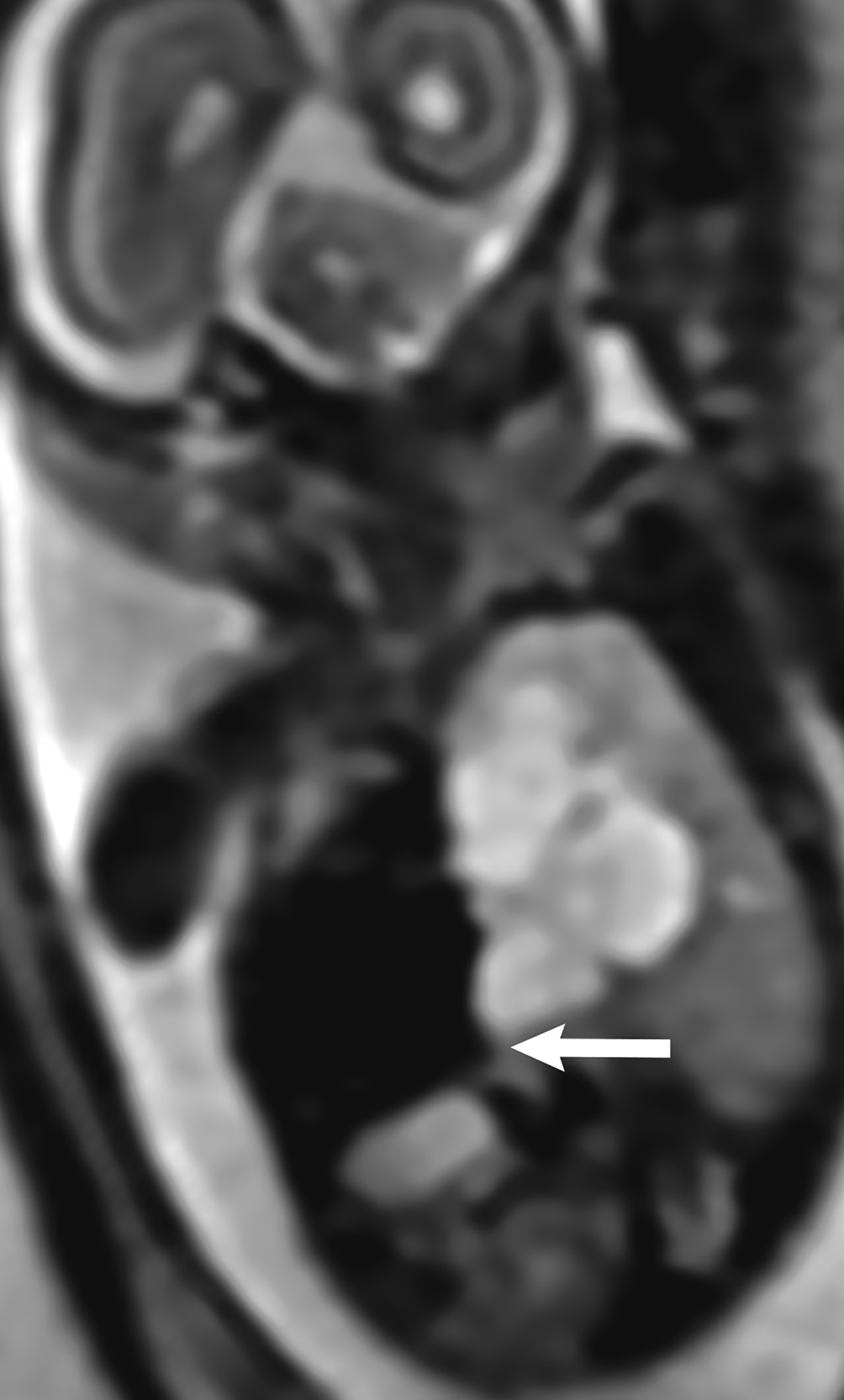
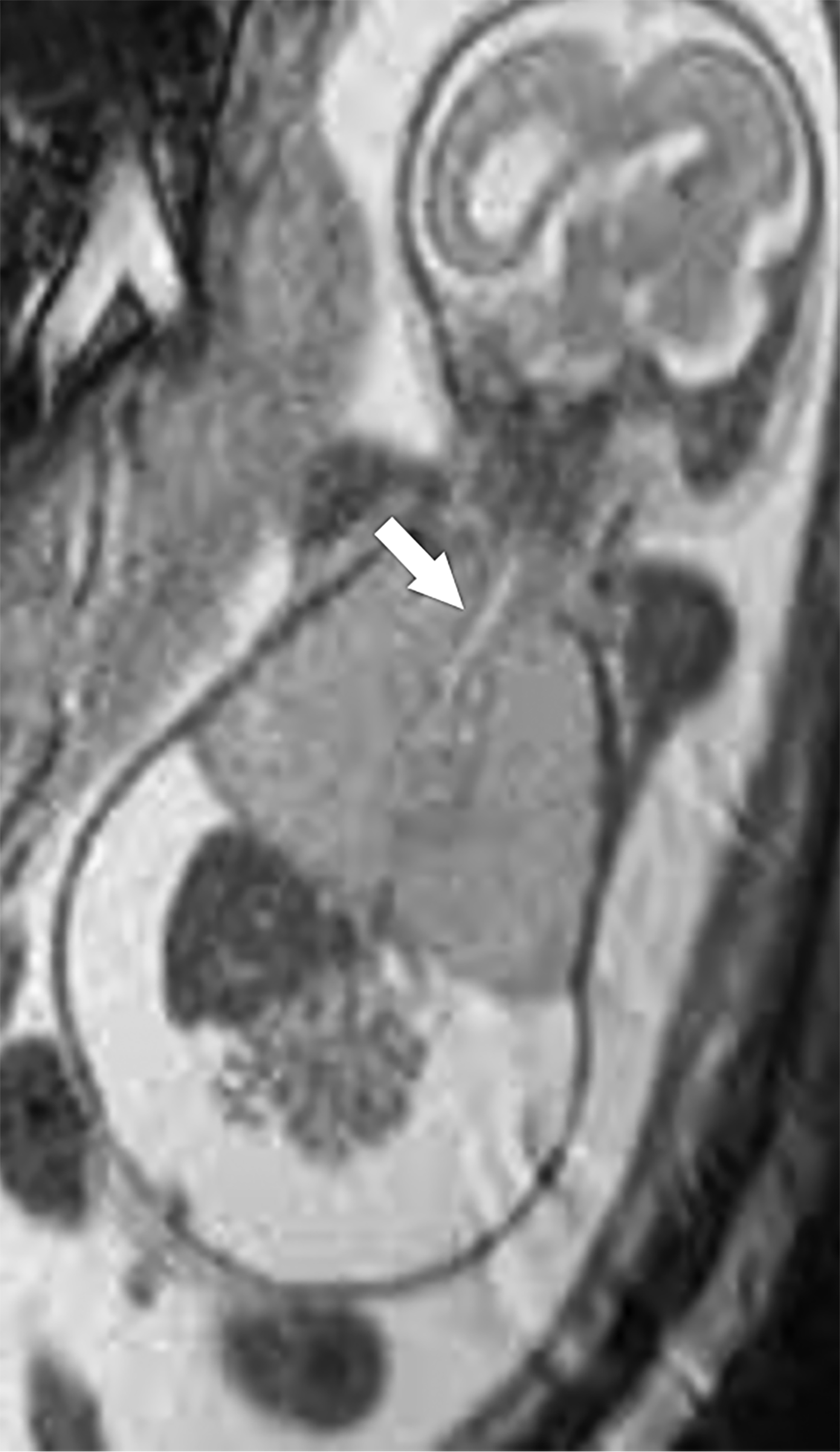
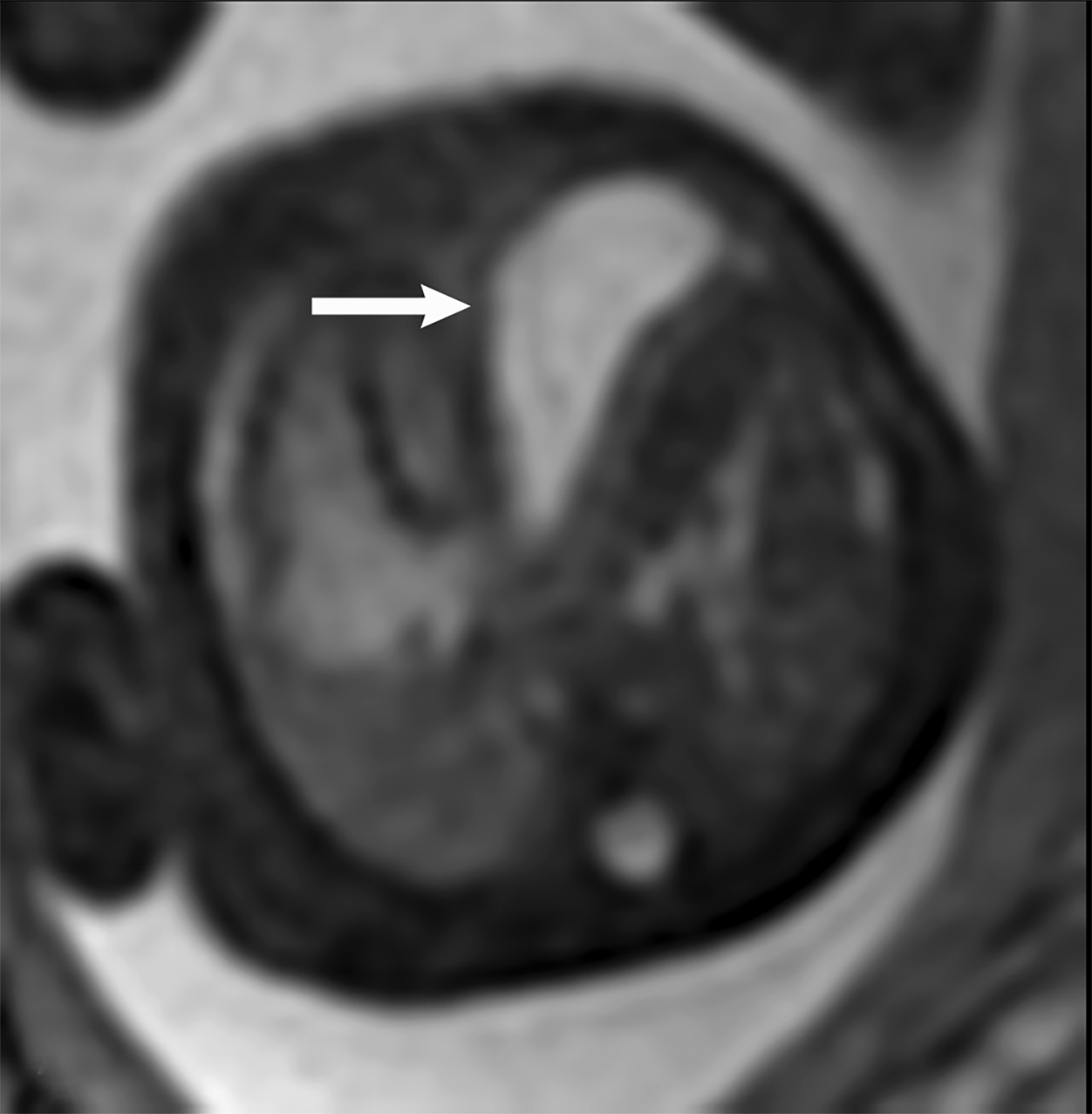
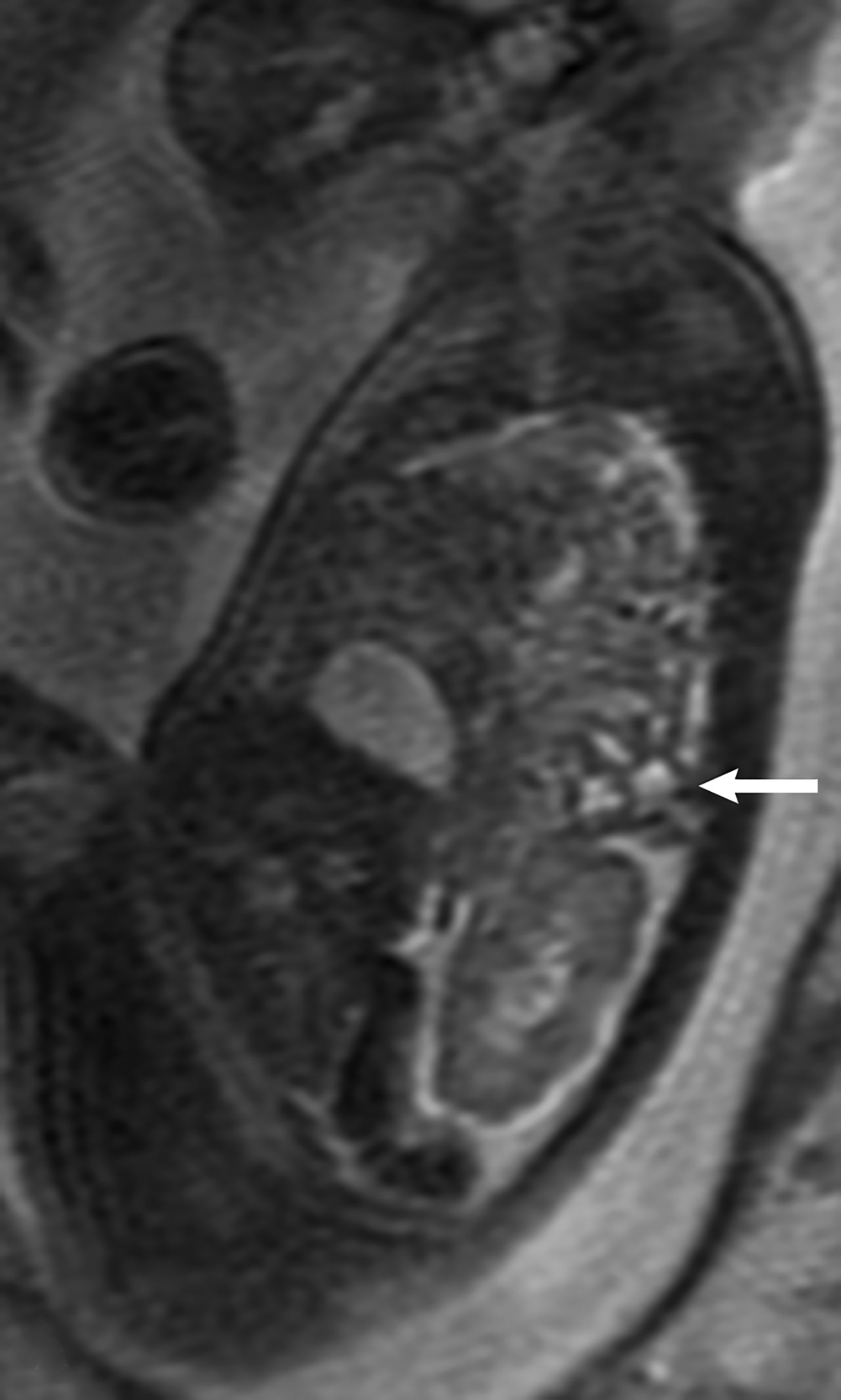
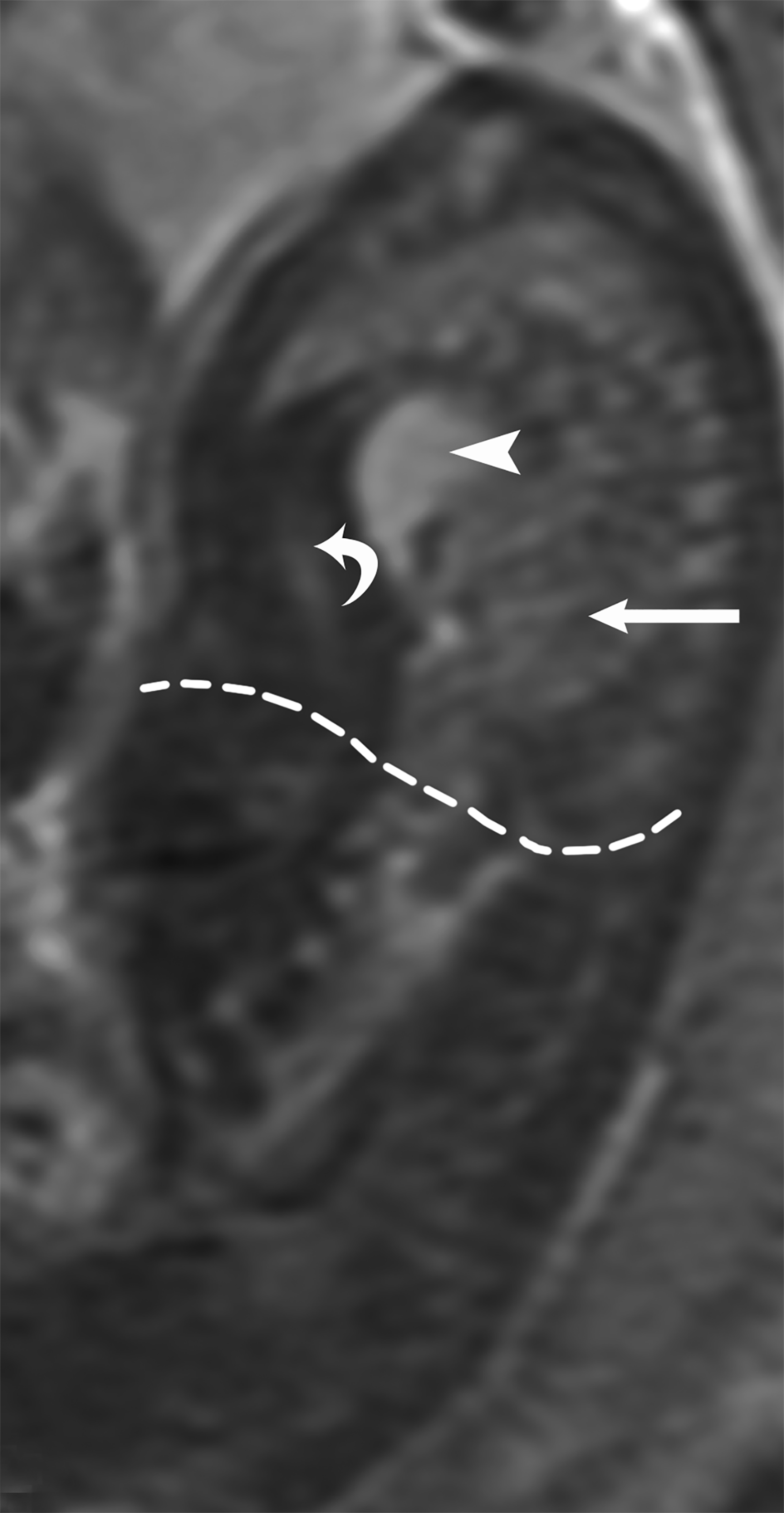
MRI can be used to corroborate ultrasound findings. For example, MRI can better discriminate the contents of the hernia given the superior soft tissue contrast and more global visualization in multiple planes (Figures 10-12). MRI is useful for more definitively identifying the side of the defect. While most CDHs are left-sided, right-sided CDHs are found in only 10 – 15 % of cases. Less than 5% of CDH cases are bilateral. MRI is also more reliable in detecting liver herniation and can calculate the percentage of liver herniation10 (Figure 13).
MRI may be used to discriminate CDH from CPAM. The bowel wall typically has low signal on T2 images, while the bowel lumen typically has high signal on T2 images. However, the cystic contents of CPAM have more homogeneously high signal on T2 images. This can be used to differentiate a bowel containing CDH from the cystic contents of CPAM (Figure 14).
Magnetic Resonance Imaging — prognostic features
Like ultrasound, MRI can also be helpful in evaluating the residual fetal lung. Ultrasound measures the cross-sectional area of the fetal lung, while MRI is useful to measure the residual lung volume. Prognosis of the CDH can be estimated in a couple of different methods, including measurement of the percent predicted lung volume (PPLV), fetal lung volume (FLV), and the observed/expected total lung volume (TLV) utilizing both the data from Meyers, et al, and Rypens, et al.11
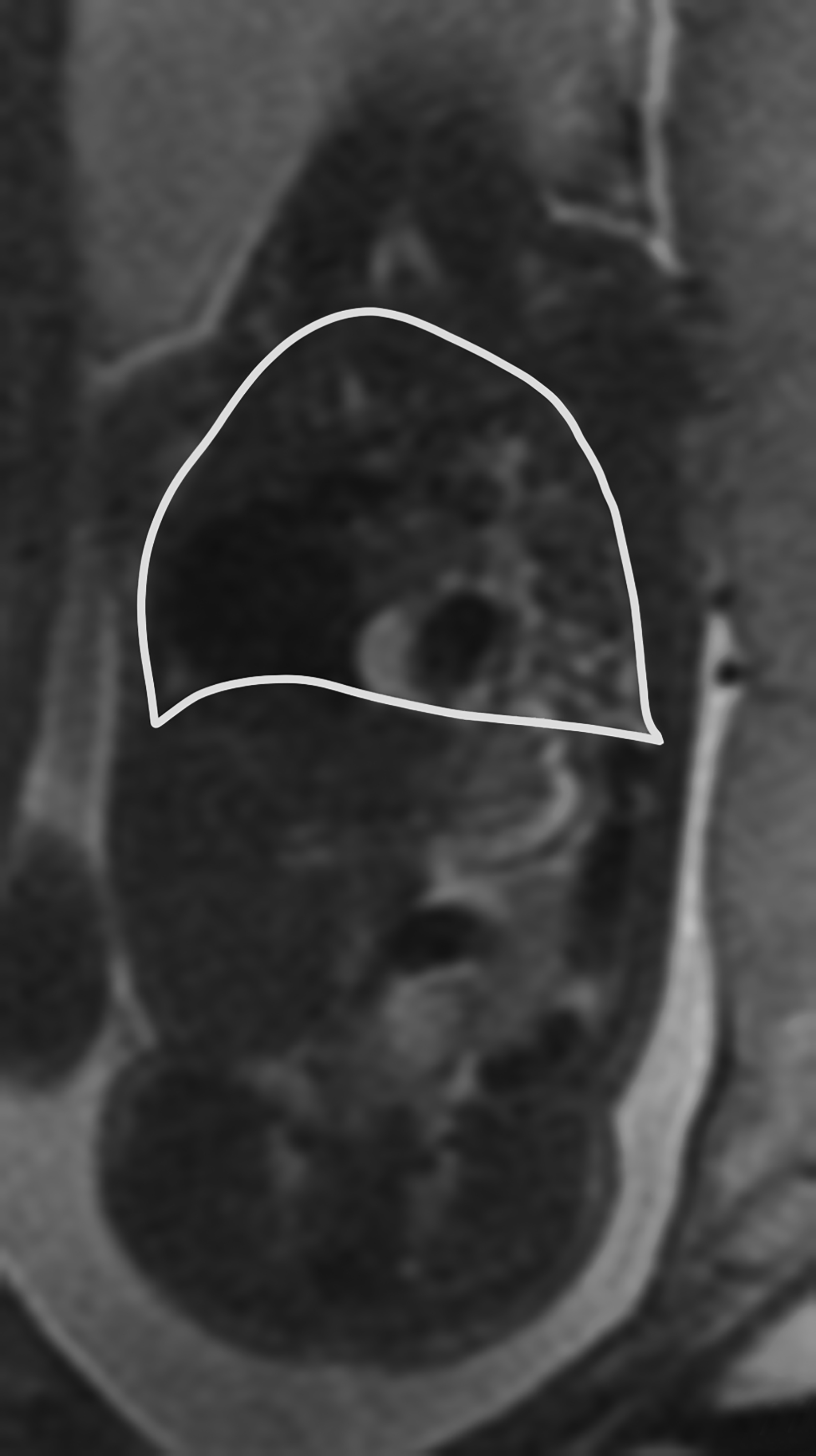
The PPLV assesses the TLV with reference to the volume of the fetus.12 To measure the PPLV, axial or coronal single shot fat spin echo (SSFSE) T2 images are used. First the thoracic volume is calculated. This is completed by taking contiguous thoracic areas and multiplying by the slice thickness.
The next step is to identify the mediastinum, which is comprised of the heart, great vessels, thymus, and central airways. These areas are likewise obtained and multiplied by the slice thickness. Then the mediastinal volume is subtracted from the thoracic volume to get the predicted lung volume (PLV) (Figure 16).12 The PPLV is calculated by dividing the actual lung volumes by the predicted lung volumes, multiplied by 100.12
PPLV =
Right lung volume + Left lung volume × 100
PLV
Percent predicted lung volumes less than 15% is associated with a poor prognosis.12
Another method for assessment of prognosis was presented by Busing et al, with measurement of the FLV.13 The volumes of each fetal lung were calculated by measuring the areas of the fetal lungs on each slice and then multiplied by the slice thickness.13 This method posits that the absolute FLV can be used as a prognostic indicator.13
The observed/expected TLV is another method of determining prognosis in the CDH population.14,15 The observed TLV of the fetus is measured by calculating the areas of the fetal lungs multiplied by the slice thickness.14,15 The expected lung volume can be derived from either the Meyers et al, or the Rypens et al, formulas. 14,15 The Rypens formula to calculate TLV is FLV = 0.0033 × (gestational age)2.86 (R2 = 0.58) with the gestational age in weeks.15 The Meyers formula to calculate TLV is 0.000865 × (gestational age)3.254 (R2 = 0.8368).14 The percentage of the observed/expected TLV would then be calculated and be utilized for prognosis.14,15
Clinical Significance
Recent advances in early diagnosis of CDH allow increasingly sophisticated prenatal management of CDH and has increased postnatal survival to greater than 70% with improvements occurring within the past 5 years. Prenatal diagnosis and imaging thus play a critical role in the management of fetuses with CDH.
Traditional management relies on antenatal glucocorticoids to promote lung development.2 The novel technique of FETO has recently been shown in a large multicenter trial to improve survival when applied at 27 – 29 weeks’ gestation for fetuses with severe CDH.16 Using this technique, the trachea is occluded in utero which causes accumulation of fluid in the lungs, thereby leading to accelerated lung growth, with the goal to minimize pulmonary hypoplasia and reduce mortality.2
Postnatally, ECMO may be required as supportive therapy in moderate and severe cases. Definitive treatment involves surgical repair of the diaphragmatic defect.
References
- Marlow J, Thomas J. A review of congenital diaphragmatic hernia. Australas J Ultrasound Med. Feb 2013;16(1):16-21.
- Kosinski P, Wielgos M. Congenital diaphragmatic hernia: pathogenesis, prenatal diagnosis and management – literature review. Ginekol Pol. 2017;88(1):24-30.
- Gupta VS, Harting MT, Lally PA, Miller CC, Hirschl RB, Davis CF, Dassinger MS 3rd, Buchmiller TL, Van Meurs KP, Yoder BA, Stewart MJ, Lally KP; Congenital Diaphragmatic Hernia Study Group. Mortality in congenital diaphragmatic hernia: a multicenter registry study of over 5000 patients Over 25 Years. Ann Surg. 2021. Doi: 10.1097/SLA.0000000000005113. Epub ahead of print. PMID: 34334632.
- Kline-Fath BM. Current advances in prenatal imaging of congenital diaphragmatic [corrected] hernia. Pediatr Radiol. 2012;42 Suppl 1: S74-90.
- Mehollin-Ray AR, Cassady CI, Cass DL, Olutoye OO. Fetal MR imaging of congenital diaphragmatic hernia. Radiographics. 2012;32(4):1067-1084.
- Recio Rodriguez M, Martinez de Vega V, Cano Alonso R, Carrascoso Arranz J, Martinez Ten P, Perez Pedregosa J. MR imaging of thoracic abnormalities in the fetus. Radiographics. Nov-Dec 2012;32(7): E305-21.
- Daltro P, Werner H, Gasparetto TD, et al. Congenital chest malformations: a multimodality approach with emphasis on fetal MR imaging. Radiographics. Mar 2010;30(2):385-395.
- Ursini WP, Ponce CC. Congenital pulmonary airway malformation. Autops Case Rep. 2018 May 2;8(2): e2018022. Doi: 10.4322/acr.2018.022. PMID: 29780758; PMCID: PMC5953188.
- perinatology.com. The Lung area to Head circumference Ratio (LHR), The Observed/expected lung-to-head ratio (o/e LHR), and The Quantitative Lung Index (QLI). Accessed January 5, 2022.
- Ruano R, Lazar DA, Cass DL, et al. Fetal lung volume and quantification of liver herniation by magnetic resonance imaging in isolated congenital diaphragmatic hernia. Ultrasound Obstet Gynecol. 2014;43(6):662-669.
- Coleman A, Phithakwatchara N, Shaaban A, Keswani S, Kline-Fath B, Kingma P, Haberman B, Lim FY. Fetal lung growth represented by longitudinal changes in MRI-derived fetal lung volume parameters predicts survival in isolated left-sided congenital diaphragmatic hernia. Prenat Diagn. 2015;35(2):160-166. Doi: 10.1002/pd.4510. Epub 2014 Nov 26. PMID: 25297802.
- Barnewolt CE, Kunisaki SM, Fauza DO, Nemes LP, Estroff JA, Jennings RW. Percent predicted lung volumes as measured on fetal magnetic resonance imaging: useful biometric parameter for risk stratification in congenital diaphragmatic hernia. J Pediatr Surg. Jan 2007;42(1):193-197.
- Busing KA, Kilian AK, Schaible T, et al. MR relative fetal lung volume in congenital diaphragmatic hernia: survival and need for extracorporeal membrane oxygenation. Radiology. 2008;248(1): 240-246.
- Meyer ML, Garcia JR, Blough KL, et al. Fetal lung volumes by MRI: normal weekly values from 18 through 38 weeks’ gestation. Am J Roentgenol. 2018; 211:432-438. Doi.org/10.2214/AJR.17.19469.
- Rypens F, Metens T, Rocourt N, et al. Fetal lung volume: estimation at MR imaging-initial results. Radiology 2001; 219(1): 236–241.
- Deprest JA, Nicolaides KH, Benachi A, et al. Randomized trial of fetal surgery for severe left diaphragmatic hernia. N Engl J Med. 2021;385:107-118. DOI: 10.1056/NEJMoa2027030.
References
Citation
A M, S S, K G, J M. Multimodality Evaluation of Fetal Congenital Diaphragmatic Hernia and Its Mimics. Appl Radiol. 2022;(6):6-16.
November 2, 2022