Neonatal juvenile granulosa cell tumor of the testis
Images

CASE SUMMARY
A baby boy born at 39 weeks’ gestation was found to have an enlarged right testicle on newborn physical exam. Subsequent ultrasound revealed a heterogeneous intratesticular mass. Serum tumor makers, including AFP, were normal for age. The patient underwent partial orchiectomy for the testicular mass. He recovered uneventfully and was discharged on day of life eight with close follow-up by urology and a primary care pediatrician.
IMAGING FINDINGS
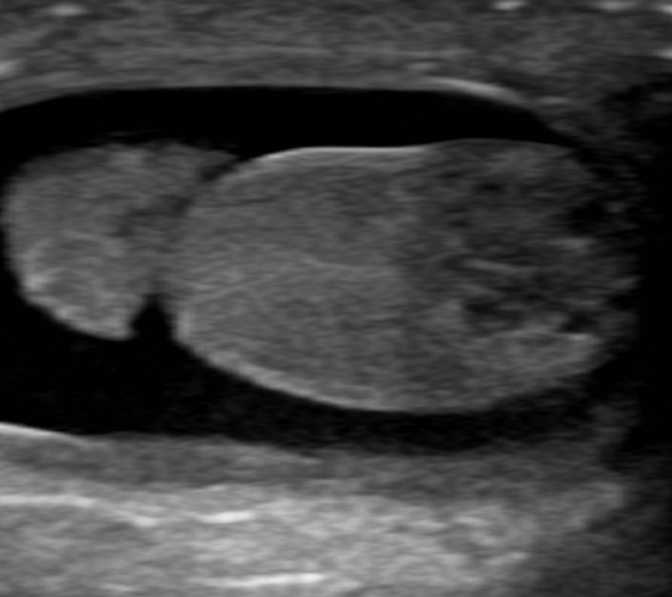
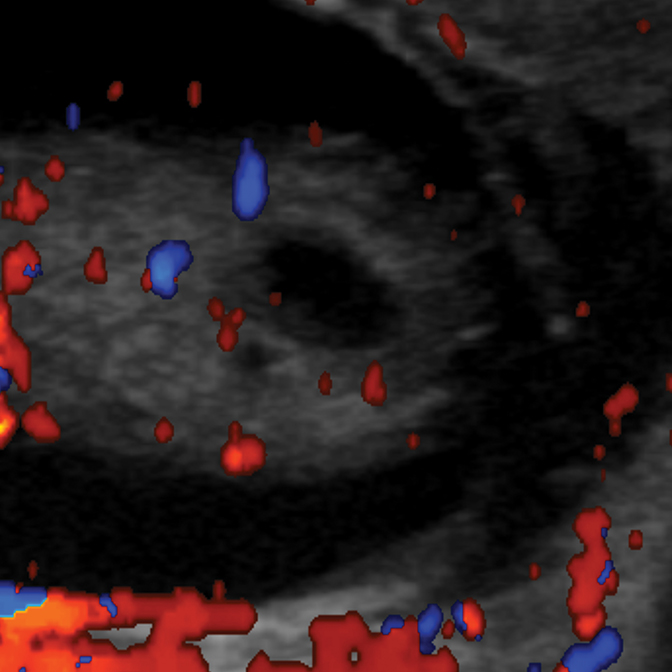
On initial ultrasound, the right testicle measured 0.8 × 0.7 × 1.3 cm in size and the left measured 0.6 × 0.8 × 0.9 cm. An approximately 7 mm mixed solid and cystic mass was identified at the inferior aspect of the right testicle (Figure 1). Color Doppler imaging detected internal blood flow within the mass (Figure 2). The left testicle was morphologically normal. Bilateral, moderate-sized hydroceles were found incidentally. Histological analysis of the tumor showed a mucinous lesion with cystic regions surrounded by granulosa cells (Figure 3).
DIAGNOSIS
The differential diagnosis includes juvenile granulosa cell tumor (JGCT), yolk sac tumor, cystic teratoma, Sertoli cell tumor, and Leydig cell tumor. Final surgical pathologic diagnosis was JGCT.
DISCUSSION
Juvenile granulosa cell tumor is a benign testicular neoplasm comprising 3 percent of primary testicular tumors in boys younger than 12 years of age.1 Although it represents the most common testicular sex cord-stromal tumor in infancy, a literature search reveals less than 100 reported cases.1 It has been postulated that JGCT originates from primitive, pluripotential gonadal stromal cells or immature Sertoli and peritubular myoid cells.2 Pathologically, the tumor contains mucin-containing follicles lined by granulosa cells which stain positive for inhibin-alpha and negative for alpha fetoprotein.3
Roughly half of JGCT present in the first few days of life, and over 90% are diagnosed by six months of age.4 In contrast, other testicular neoplasms in the prepubertal population present later in life. The malignant yolk sac tumor presents at a median age of 16 months, and teratomas and Sertoli cell tumors present at median ages of 13 months and 6 months, respectively.1 It is highly unusual for Leydig cell tumors to present prior to 2 years of age.1 Enlargement of a scrotal testis is the presenting manifestation in most JGCT, while a minority of cases are found during work up for ambiguous genitalia, undescended testicles, or testicular torsion.4,5 An association with chromosomal anomalies, including structural abnormalities of the Y chromosome, has been documented.5 Characteristically, patients do not have evidence of associated endocrine abnormalities. In contrast, Leydig cell tumors may be hormonally active in the prepubertal testis resulting in sexual precocity.4 Serum alpha fetoprotein levels in JGCT are normal for patient age, a finding which reliably excludes a yolk sac tumor.2,3
Initial imaging of JGCT is usually accomplished by ultrasound, which shows a predominantly cystic mass or partly cystic lesion with prominent solid components. The tumors range in size from 0.8 cm to 5.0 cm and usually occupy most of the testis with residual parenchyma confined to a thin rim.4 Often the testicular parenchyma is completely replaced by multiseptated, cystic structures. The rim of the cystic mass may appear hypervascular on color Doppler imaging6; however, this is not universally present.7 Rare cases of neonatal abdominal juvenile granulosa cell tumors originating from from undescended testicles appear as well-circumscribed, multicystic and multiseptated masses. MRI demonstrates expected increased T2 signal within the cystic components.8
Historically most cases of testicular JGCT have been treated with unilateral orchiectomy; however, testis-sparing enucleation has been performed with increasing frequency. To the extent that follow-up has been available for patients following resection, no cases of recurrence or metastasis have been reported with either approach.3,4,9 This benign course is somewhat unexpected, as histologically JCGT often show locally aggressive and infiltrative growth patterns, dense cellularity, and considerable mitotic activity.9 Given the favorable long-term outcome, enucleation has been advocated as the treatment of choice provided that serum AFP is normal.3
CONCLUSION
Juvenile granulosa cell tumor is a rare testicular sex cord-stromal neoplasm in infancy. The vast majority of cases present prior to 6 months of age, while other pediatric testicular tumors present later in life. The tumors do not cause precocious puberty or abnormally elevated serum AFP levels. Ultrasound findings include a cystic or partly cystic mass with solid components. When large, the tumors may appear as multiseptated cystic lesions replacing most of the affected testicle. The behavior of JGCT is clinically benign, and definitive treatment may be accomplished by orchiectomy or testis-sparing enucleation.
REFERENCES
- Ross JH, Rybicki L, Kay R. Clinical behavior and a contemporary management algorithm for prepubertal testis tumors: a summary of the Prepubertal Testis Tumor Registry. The Journal of Urology. 2002;168:1675-1679.
- Groisman GM, Dische MR, Fine EM, Unger PD. Juvenile granulosa cell tumor of the testis: a comparative immunohistochemical study with normal infantile gonads. Fetal & Pediatric Pathology. 1993;13:389-400.
- Shukla AR, Huff DS, Canning DA, et al. Juvenile granulosa cell tumor of the testis: contemporary clinical management and pathological diagnosis. The Journal of Urology. 2004;171:1900-1902.
- Lawrence WD, Young RH, Scully RE. Juvenile granulosa cell tumor of the infantile testis: A report of 14 cases. The American Journal of Surgical Pathology. 1985;9:87-94.
- Nistal M, Redondo E, Paniagua R. Juvenile granulosa cell tumor of the testis. Archives of Pathology & Laboratory Medicine. 1988;112:1129-1132.
- Zugor V, Labanaris AP, Witt J, et al. Congenital juvenile granulosa cell tumor of the testis in newborns. Anticancer Research. 2010;30:1731-1734.
- Couture J, Bolduc S. A rare testicular solid mass in children: juvenile granulosa cell tumour of testis. Canadian Urological Association Journal. 2012;6:E101.
- Leylek AM, Kane RA. Juvenile granulosa cell tumor of the testis. Ultrasound Quarterly. 2014;30:219-220.
- Harms D, Kock LR. Testicular juvenile granulosa cell and Sertoli cell tumours: a clinicopathological study of 29 cases from the Kiel Paediatric Tumour Registry. Virchows Archiv. 1997;430:301-309.
Citation
S L, P K. Neonatal juvenile granulosa cell tumor of the testis. Appl Radiol. 2016;(7):32A-32B.
July 2, 2016