Neurological and Orbital Involvement of Systemic IgG4-related Sclerosing Disease
Images
Brought to you by Guerbet.
Case Summary
A 70-year-old male presented to neurology with a complaint of persistent tinnitus. Contrast-enhanced magnetic resonance imaging (CEMRI) of the brain revealed enhancing lesions within the pons, left middle cerebellar peduncle, right hypothalamus, and left hippocampal regions. The diagnostic work-up included an abdominal computed tomography (CT) study, which revealed significant retroperitoneal stranding, for which the patient underwent CT-guided biopsy. Histopathology demonstrated features suggestive of Immunoglobulin G4 (IgG4)-related retroperitoneal fibrosis. The patient began a 6-month taper of oral prednisone but eventually experienced abnormal visual changes. A follow-up CEMRI of the brain and orbits revealed new left retrobulbar involvement. Subsequently, rituximab was added to his management regimen and MRI was repeated at 6-month intervals. Asymptomatic periods were punctuated by periods of seizure activity and memory loss, and mycophenolate and dexamethasone were added to the patient’s therapy. Due to concerns of disease progression of the ocular lesion, neurosurgical intervention was performed. Today, the patient remains clinically stable on dexamethasone and continues to follow-up with hematological and MRI surveillance.
Diagnosis
IgG4-related sclerosing disease of the central nervous systems (CNS) with orbital involvement.
Imaging Findings
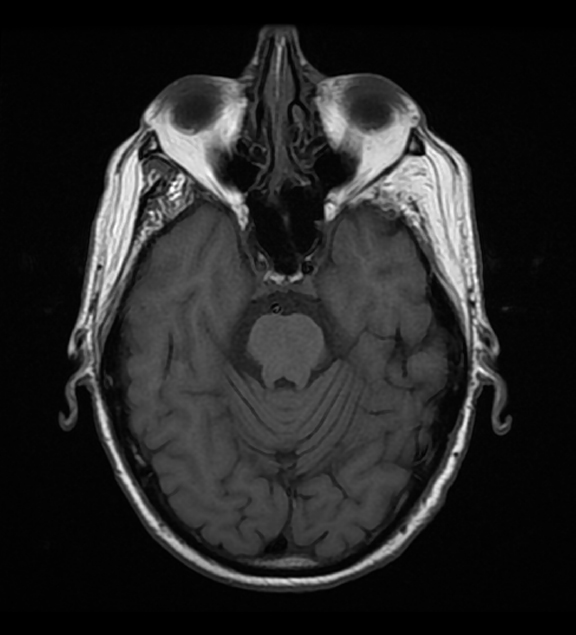
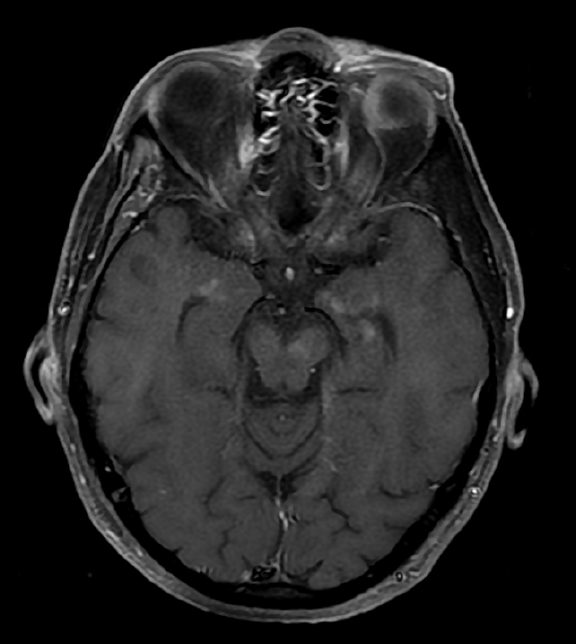
Initial Dotarem® (gadolinium-based contrast) enhanced brain MRI (Figure 1) demonstrated evidence of postcontrast, T1 hyper-enhancing signal abnormality in the pontine tegmentum and tectum and extending from the pons into the lower midbrain. The lesion was central and slightly lateralized to the right. No other abnormal enhancement was observed.
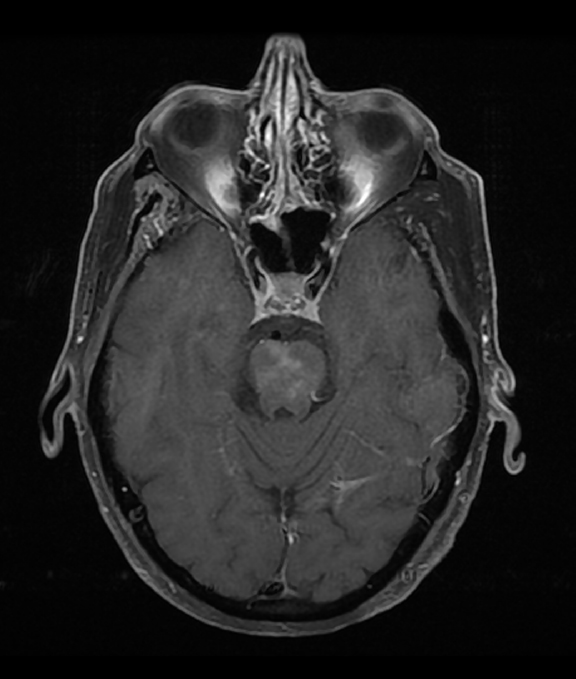
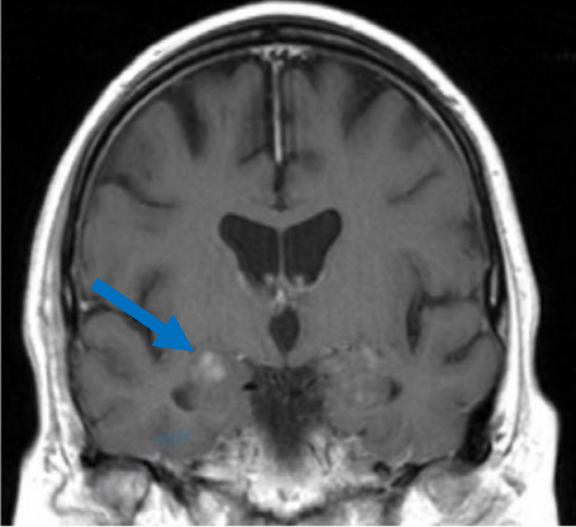
A CEMRI scan of the brain (Figure 2) performed 3 years after the initial MRI, showed slight increase in enhancement of the pons, as well as medial temporal lobe involvement. The scan also revealed a new left ocular globe and optic nerve enhancement.
Discussion
Immunoglobulin G4- related disease (IgG4-RD) is a chronic, autoimmune, fibro-inflammatory disorder first recognized as a unified entity early in the 20th century in Japan.1 Often misdiagnosed as malignancy or infection, the disease can affect any organ system, including the nervous system. While orbital disease remains a rare complication on the IgG4-RD spectrum, a Japanese study found that it comprises about 20 percent of all the infiltrative processes (malignancy or inflammation) that can affect the orbital tissue and structures.2 Regardless of the organ system, the hallmark of IgG4-RD is a subacute tumor-like swelling of the involved organ3. To date, the precise prevalence of IgG4-RD disease remains undetermined, although an outdated estimate in Japan reported a prevalence of approximately 1.08 per 100,000 of population.4 IgG4-RD demographics had a minor, albeit noteworthy, predilection to elderly males in almost all categories of the disease except for head, neck, and neurological involvements, which were more common among females5.
From a histopathological standpoint, IgG4-RD lesions share a common triad of dense, IgG4-producing plasma cell infiltrates, obliterative phlebitis, and ‘storiform’ fibrosis.6 The exact pathogenesis of IgG4-RD remains under investigation but likely reflects an antigen-driven inflammatory process. Both B-cell (blasmablast) and T-cell (T-follicular helper cells and CD4+ cytotoxic T-lymphocytes (CTL)) linage are believed to play the main role in IgG4-RD pathogenesis5. Though not confirmed, recent evidence points to the role of autoantigens such as annexin A11, galectin 3, and laminin 111 in initiating the immune cascade.7 The concentration of plasmablasts in the serum of a patient with IgG4-RD appears to correlate well with disease activity10. This suggests an interaction of plasmablasts and plasma cells with the CTL and the T-helper lymphocytes, the latter of which control IgG4 production and are abundant in IgG4-RD involved tissue.10
The constellation of autoimmune manifestations of IgG4-RD are often subacute, with tissue involvements often found incidentally on imaging or progressing over months or years before becoming detectable. Most patients present to ambulatory settings with nonspecific complaints.11 Systemic disease may present acutely with constitutional symptoms, although fever is not typical.11 Neurological manifestations result either from direct tissue infiltration or from space-occupying lesion compression. The most common IgG4-RD neurological involvements manifest in pachymeningitis and hypophysitis (pituitary gland and stalk), along with brain parenchyma and peripheral nervous system involvement. Nearly 50% of patients with IgG4-related neurological disease present with orbital and perineural disease.11 Clinical manifestations of IgG4-related orbital disease depend on the location and the orbital structures involved, with infra-orbital nerve involvement most suggestive of IgG4- related disease.11
Multiple attempts to construct diagnostic criteria for IgG4-RD have been published; however, several challenges with the proposed criteria have been identified in clinical practice.12 Nonetheless, conclusive diagnosis of IgG4-RD classically requires the correlation of clinical manifestations with MRI findings, serology, and tissue biopsy.13 On neurologic MRI, IgG4-related lesions demonstrate both patchy and confluent postcontrast hyper-enhancement and appear hypointense on T2-weighted sequencing due to fibrosis. In the orbit, a tumefactive appearance of abnormal signal and enhancement is often seen as an “orbital pseudotumor.”14
Early diagnosis and intervention are key determinants of IgG4-RD prognosis. Initial therapy is aimed at inducing remission and is often indicated in the setting of debilitating active symptomatic disease or asymptomatic evidence of progressive disease.15 Glucocorticoid monotherapy is the mainstay of therapy for systemic disease, although patients with multi-organ, neurological, or orbital disease may benefit from the addition of a biologic agent such as rituximab, which derives its therapeutic function by depleting B cells.16 Owing to the conspicuity of the lesions, particularly on post-contrast imaging, MRI is useful for monitoring therapy response and disease progression.
Conclusion
We report a case of systemic sclerotic IgG4-RD, with CNS and orbital involvement, in which CEMRI is integral to diagnosis and to surveillance. Diagnosis of IgG4-RD is multifactorial, but some imaging findings have been well described and lend a degree of specificity. Inflammatory modulator therapy is most effective during the proliferative and inflammatory stages of the disease. Glucocorticoids are usually effective at inducing remission. Patients with evidence of recurrence or failure of therapy, may benefit from a combination of glucocorticoids and rituximab. Given the limited pathogenic understanding of IgG4, further research into IgG4-RD imaging and treatment is warranted and will likely be of value.
References
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001; 344: 732–38.
- Aihara Y, Azumi A, Furuta M. et al. A prevalence study of IgG4-related ophthalmic disease in Japan. Jpn J Ophthalmol. 2013; 57: 573–79.
- Wallace ZS, Khosroshahi A, Jakobiec FA, Deshpande V, Hatton MP, Ritter J, Ferry JA, Stone JH. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, including orbital myositis, and trigeminal nerve involvement. Surv Ophthalmol. 2012;57(1):26.
- Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012; Feb;22(1):1-14.
- Baptista B, Casian A, Gunawardena H, D’Cruz D, & Rice CM. Neurological manifestation of IgG4-related disease. Curr Treat Opinions Neurol. 2017; 19(4): 14.
- Umehara H, Okazaki K, Nakamura S, et al. Current approach to the diagnosis of IgG4-related diseae- combination of comprehensive diagnostic and organ-specific criteria. Mod Rheumatol. 2017; 27(3): 381-391
- Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, Allard-Chamard H, Wallace Z, Montesi SB, Kreuzer J, Haas W, Stone JH, Pillai S. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol. 2019;143(2):736.
- Johnson J & Allen JE. IgG4-related disease in the head and neck. Curr Opin Otolarynfol Head Neck Surg. 2018; 26(6): 403-408.
- Nizar A-H, Toubi E. IgG4-related disease: case report and literature review. Auto Immune Highlights. 2015; 6(1-2):7- 15.
- Stone JH. IgG4-related disease: pathophysiologic insights drive emerging treatment approaches. Clin Exp Rheumatol. 2016;34(4 Suppl 98):66–8.
- Abdel Razek MA, Venna N, Stone JH. IgG4-related disease of the central and peripheral nervous system. Lancet Neurol. 2018; 17:183-92.
- Sato Y, Notohara K, Kojima M, Takata K, Masaki Y, & Yoshino T. IgG4-related disease: historical Overview and pathology of hematological disorders. Pathol In. 2010; 60(4)247-58.
- Babptista B, Casian A, Gunawadena H, D’Cruz D, & Rice CM. Neurological Manifestations of IgG4-Related Disease. Curr Treat Options Neurol. 2017; 19:14.
- Soussan JB, Deschamps R, Sadik JC, et al. Infraorbital nerve involvement on magnetic resonance imaging in European patients with IgG4-related ophthalmic disease: a specific sign. Eur Radiol.2016; 27: 1335–43.
- Zhang W, & Stone JH. Management of IgG4-related disease. Lancet Rheumatol. 2019; 1:e55.
- Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015; 67: 1688–99.
Citation
Neurological and Orbital Involvement of Systemic IgG4-related Sclerosing Disease. Appl Radiol.
February 26, 2021