Radiological Case: Ollier’s disease
Images
CASE SUMMARY
The patient was an 11-year-old girl with a longstanding history of musculoskeletal deformities of her extremities. The condition was first discovered at birth, and she had been closely followed by multiple providers at several institutions to document the progression of her disease. She was capable of performing routine daily activities with little assistance, except for the use of a wheelchair after walking significant distances. The patient exhibited no recent fevers, chills, weakness or bone pain out of proportion to her baseline; however, a nuclear medicine scan in conjunction with plain radiographs was requested to document the extent of her disease due to fears of malignant degeneration of her condition.
IMAGING FINDINGS
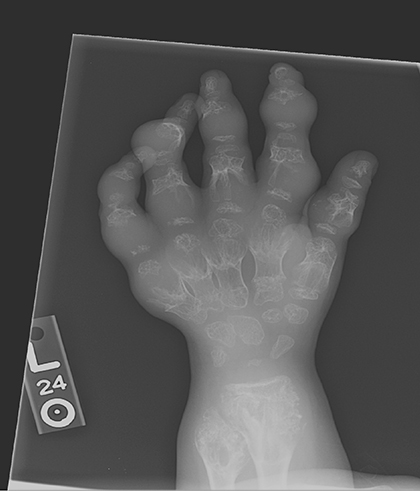
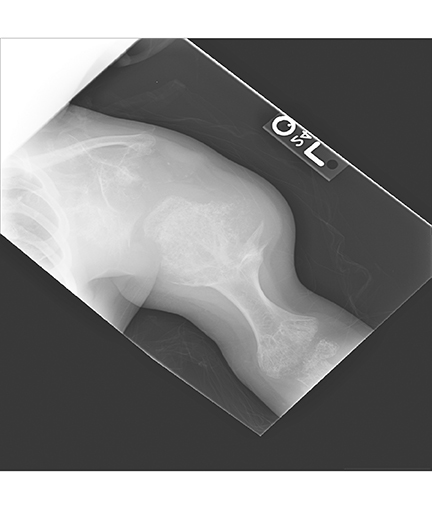
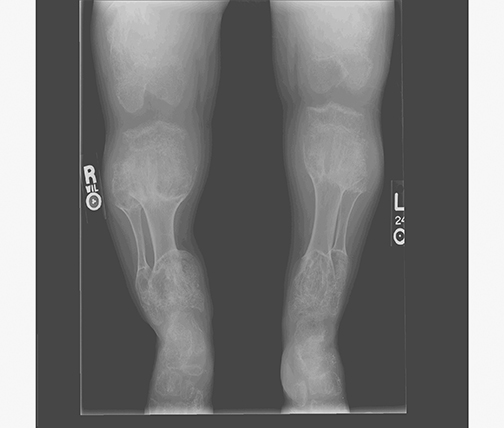
AP views of the left hand, left shoulder and bilateral knees demonstrated numerous expansile, mixed lytic and sclerotic metasphyseal lesions of the tubular bones, as well as the scapula (Figures 1-3). Although soft-tissue contour was distorted by underlying skeletal deformities, there was no evidence of soft-tissue mass or calcifications.
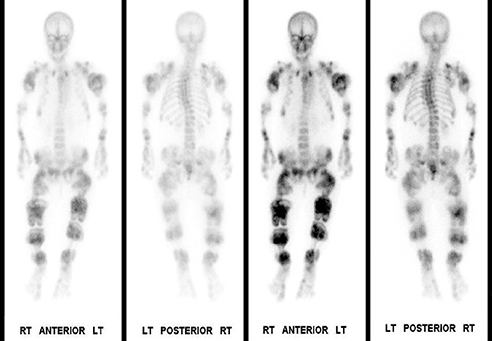
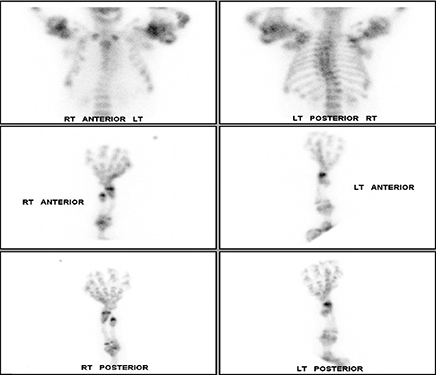
Whole-body bone scintigraphy following administration of 19.63 mCi Tc-99m HDP showed multiple lobulated and expansile areas of intense radiopharmaceutical accumulation in the axial and appendicular skeleton, more pronounced in the latter, particularly in the lower extremities and the proximal humeri assuming the appearance of round rubber rings as in the “Michelin Man” (Figure 4A,B). The increased activity was suggestive of metabolic activity in these lesions that were probably actively calcifying. Additionally, there was severe scoliosis of the dorsolumbar spine. No abnormal soft-tissue uptake was noted.
DIAGNOSIS
Ollier’s disease
DISCUSSION
Ollier’s disease is a nonhereditary skeletal disorder that typically manifests in childhood. First described by Ollier in 1899, this rare condition has been documented only slightly more than 100 times in the 30 years prior to 1998.1,2 The condition manifests as multiple, subperiostial, cartilaginous deposits (enchondromas), most often located within the long and flat bones, such as the femur, tibia, and pelvis, as well as the small bones of the hands and feet. The lesions have been noted to be distributed either unilaterally or bilaterally, with a slight predilection for unilateral occurrence.3 Clinically, patients usually present during the first decade of life, exhibiting arthralgias, palpable bony masses, asymmetry, and/or extremity shortening with or without pathological fractures. Disruption of the physis occurs often, leading to permanent growth defects and deformation throughout the patient’s life. Although the disease typically occurs early in life, these lesions most often stabilize in size over time with occasionally reported regression later during adulthood. Ollier’s disease also confers an increased risk of malignancy, with transformation of the enchondromas into chondrosarcomas being noted, most often around age 40.4 Occurrence of this phenomenon is still debatable, with ranges listed from 5-50%.3,5 Treatment generally consists of bone curettage and grafting. However, other experimental methods, such as external fixation with an Ilizarov device, have been employed with some success.5 Prognosis depends on extent of the disease, incidence of pathologic fractures, malignant transformation, and degree of deformity.
Ollier’s disease is generally diagnosed via several imaging modalities, including radiography, CT, MRI, and nuclear medicine. Generally radiography is the first diagnostic modality utilized, due to its low cost and good probability of lesion detection; CT is often used in conjunction with radiography. Lesions are typically expansile, displaying a pattern of calcifications described as central “rings and arcs” throughout the affected bones without cortical penetration. Transformation into chondrosarcoma generally is indicated when cortical breakthrough is noted along with soft-tissue involvement. MRI is often employed when diagnostic certainty cannot be made with either radiography or CT. Generally the lesions appear similar to other hyaline cartilage foci, with high signal intensity foci on T2-weighted images and low intensity demonstrated on T1 images.6 Finally, disease extent can often be documented with technetium-99m MDP radionucleotide bone scintigraphy. Radiopharmaceutical uptake can vary greatly depending upon local reactive bone formation, ossification of the enchondromas, and local hyperemia.7 However, the imaging manifestations of malignant degeneration are often nonspecific and clinical findings of new or progressive pain are considered to be the most sensitive findings suggesting transformation to chondrosarcoma. Beyond this, biopsy remains the final determination of such a process.
CONCLUSION
Ollier’s disease, as represented in this case, is readily identified due to classic radiographic and scintigraphic manifestations. However, detecting malignant degeneration continues to present a challenge in diagnostic imaging. Determination of malignant transformation continues to rely primarily on clinical presentation of pain and rapid enlargement of the tumors. In addition, a pathological specimen is needed to definitively diagnose malignant transformation despite advances in new diagnostic imaging techniques.
REFERENCES
- Shapiro, F. et al. Ollier’s disease: An assessment of angular deformity, shortening, and pathological fracture in twenty-one patients. J Bone Joint Surg. 1982;(64A):95-103.
- Hasbini A, Lartigau E, Le Pechoux C, et al. Les chondrosarcomes sur maladie d’Ollier. A propos de deux cas et revue de la literature. Cancer/Radiotherapie. 1998;(2):387-391.
- Kaya, H, Komeck, H, Cerci, S, et al. Bilateral symmetrical Ollier disease and Tc-99m MDP Bone scintigraphy. Clin Nuc Med. 2004;29(7):456.
- Resnick, D, et al. Diagnosis of Bone and Joint Disorders. 4th Ed., WB Saunders Co 2002:4502
- Jesus-Garcia R, Bongiovanni JC, Korukian M, et al. Use of ilizarov external fixator in the treatment of patients with Ollier’s disease. Clin Ortho Related Res. 2001.382:82-86.
- Chew FS, Maldjian C. Enchondroma and enchondromatosis. www.emedicine.com, 2005.
- Henkin R, Boles M, Dillehay G, et al. Nuclear Medicine, Volume II. St. Louis, MO: Mosby 1996:1707.