Work-up and Imaging of Congenital Lung Malformations
Images




Recent years have seen major advances in pathological and radiological approaches to congenital lung malformations (CLM) with secondary subsequent re-evaluation of management options. These advances have stemmed from newer theories of in utero malformation induction and higher-resolution prenatal and postnatal imaging. Furthermore, studies have begun to clarify the potential relationship between some congenital malformations and rare pulmonary malignancies. This review will explore these issues in relation to the more common CLMs. Accepting that the literature is still controversial with regard to subsequent management, we present a “how we do it” approach.
Embryological Basis of Congenital Lung Malformations
In previous years, the various parenchymal malformations were evaluated and treated as isolated entities. With time, the introduction of widespread prenatal sonography resulted in prenatal diagnoses that proved radiologically and pathologically inaccurate after resection. These inaccuracies were related to both size and content of the CLM. Many lesions demonstrated significant regression over time. As well, lesions were beginning to be recognized as having “hybrid;” ie, mixed, pathological findings, usually a combination of cystic change, suggestive of a congenital pulmonary airway malformation (CPAM), and a systemic arterial supply, suggestive of pulmonary sequestration.1-5
A landmark paper by Langston et al,6 however, brought a new and somewhat unifying approach to the potential in utero mechanism by which many of these malformations may occur. Pediatric pathologists have observed the presence of an atretic bronchus in many different types of CLMs. As such, in utero airway obstruction has been suggested as a uniting cause of malformation induction in some of the most common entities, including congenital lobar emphysema/overinflation (CLOE), CPAM, and bronchial atresia. This results in pathological findings of overlapping CLM elements within single lesions. The classification scheme proposed by Langston et al suggests that most lesions develop as sequelae of in utero airway obstruction, resulting in tracheobronchial and pulmonary mal-development. The spectrum of pulmonary anomalies that are manifested depends on the level, timing, and degree of bronchial obstruction. For example, the clinical and radiological manifestations of uncomplicated bronchial atresia represent an atresia that happened relatively late in utero, after the tracheobronchial tree and associated lung had already matured to some extent. In contrast, an atresia that occurs earlier in utero may result in a more disorganized, dysplastic lobe peripheral to the associated atretic bronchus, as seen in most cases of CPAM, where the dysplasia is manifested as a multicystic malformation.1,6 The concept is similar to the formation of a multicystic dysplastic kidney secondary to disordered formation of the renal pelvis.
Imaging and Clinical Presentation of Congenital Lung Malformations
Bronchial Atresia
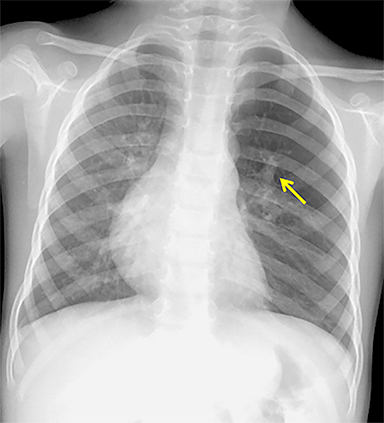
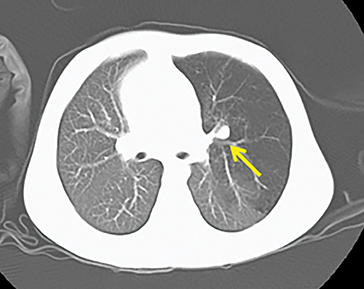
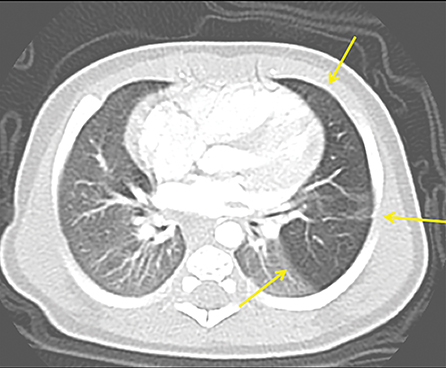
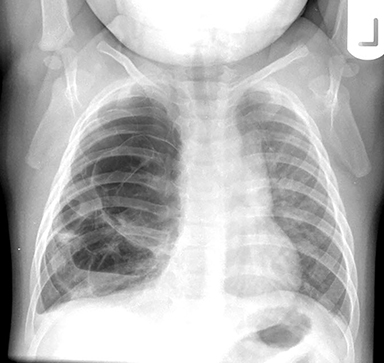
The imaging appearance of isolated bronchial atresia is a hyperlucent lobe, most commonly the left upper lobe, with central perihilar “finger-shaped” tubular opacities representing inspissated mucous distal to the atretic segment (Figure 1). Hyperinflated lung seen distal to the atretic segment is secondary to air filling through collateral pathways. This isolated manifestation of bronchial atresia supposes the atresia occurred later in utero.
Patients are usually minimally symptomatic, if at all, but they can present with respiratory distress or recurrent infection. The etiology commonly proposed for the obliteration of the connection between primitive bronchial cells and the bronchial bud tip is a focal vascular insult, similar to bowel atresias, resulting in obliteration of the proximal airway and segmental or subsegmental bronchial obstruction.6,7
CLOE
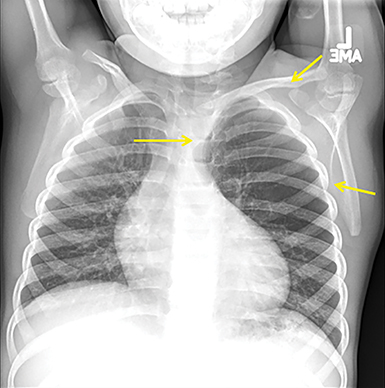
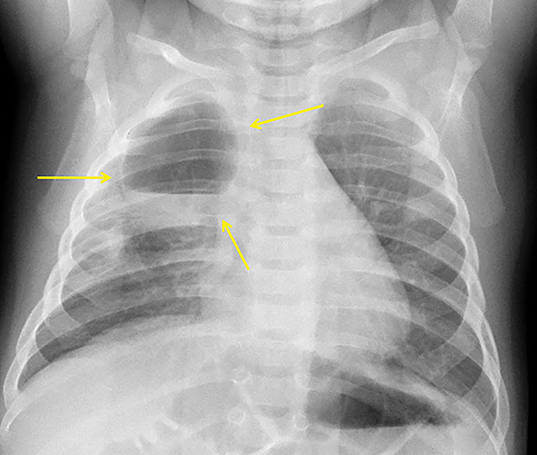
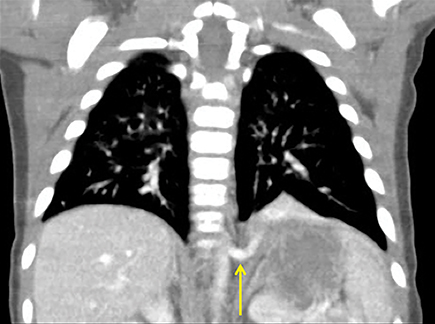
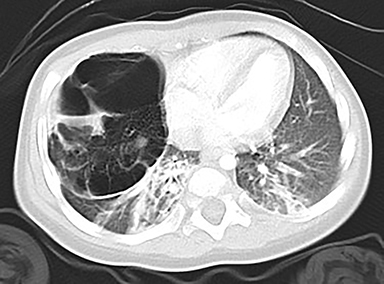
In the early neonatal period, CLOE may present on radiographs as a focal opacity secondary to retained fetal fluid. Aeration progresses within days as the fluid is cleared, followed by subsequent overinflation from air trapping and collateral air drift. The left upper and right middle lobes are most commonly affected. Progressive overinflation may result in compression of the contralateral lung and mediastinal shift (Figure 2).
CLOE can result from intrinsic or extrinsic narrowing of the bronchial lumen, with subsequent lobar hyperinflation. Approximately half of these malformations are attributable to demonstrable anomalous formation of the feeding bronchus to the affected lobe, usually the left upper or right middle lobes. Resultant bronchomalacia or bronchostenosis occurs, whether from a primary stenosis or secondary to an adjacent lesion, such as a centrally positioned bronchogenic cyst. Histologically, the overinflation can be due to overdistention of an otherwise anatomically normal lobe, or overinflation of a lobe due to an increased number of normally inflated alveoli, producing the so-called “polyalveolar lobe.”8
CPAM
These malformations are the most common congenital lung malformations and are most often identified at prenatal sonography. They are suspected to arise from dysplastic overgrowth of normal terminal bronchiolar tissue following early in utero bronchial obstruction, as described previously.
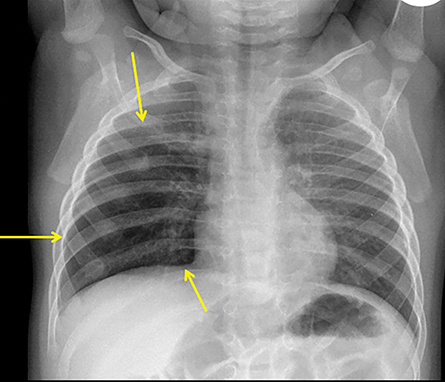
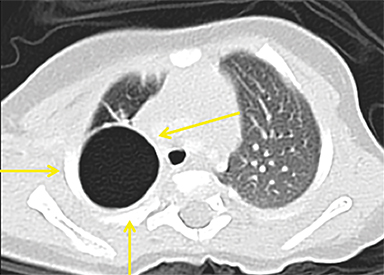
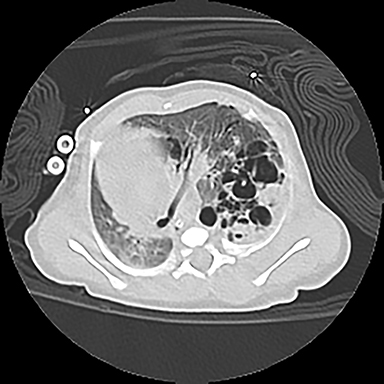
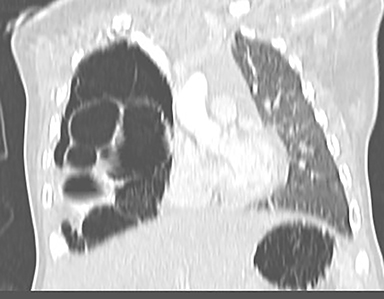
CPAMs appear as foci of pulmonary parenchymal cystic dysplasia of varying size and compression of regional structures. Many children may be asymptomatic, but infants can present with significant respiratory distress, usually due to mass effect. Older children, similar to that of younger children with other CLMs, can present with recurrent infections. Stocker et al9 initially devised a classification system for CPAMs in which lesion were classified according to cyst size and tissue origin: Type 1 lesions were greater than 2 cm and of bronchial/bronchiolar origin; type 2 ranged from 0.5 to 2 cm in size and of bronchiolar origin; and type 3 were <0.5 cm, solid-appearing lesions comprising multiple, fluid-filled cysts of bronchiolar/alveolar origin (Figure 3). This classification system was widely accepted, and it remains the basis of most practical approaches to these lesions. More recently, Stocker10 further revised the system to include types 0-4 with several subtypes: Type 0 manifests as small lungs with severe acinar dysgenesis and is incompatible with life, and type 4 is of distal acinar origin with large peripheral cysts and mass effect, with possible crossover between type 4 lesions and pleuropulmonary blastoma.
Pulmonary Sequestrations
Pulmonary sequestrations are the second-most common congenital lung malformations. They are characterized by dysplastic pulmonary tissue supplied by the systemic arterial system, with no normal connection to the tracheobronchial tree.
Sequestrations can be classified as extralobar or intralobar. Extralobar sequestrations have their own pleural investment, are most commonly seen in the posterior aspect of the left lower lobe, are uncommonly symptomatic, and more frequently demonstrate systemic venous drainage. Intralobar sequestrations share the pleura with the remainder of the ipsilateral lung, more frequently have pulmonary venous drainage, and more commonly demonstrate visible cystic change.11- 12
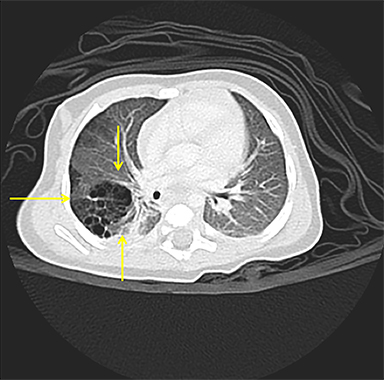
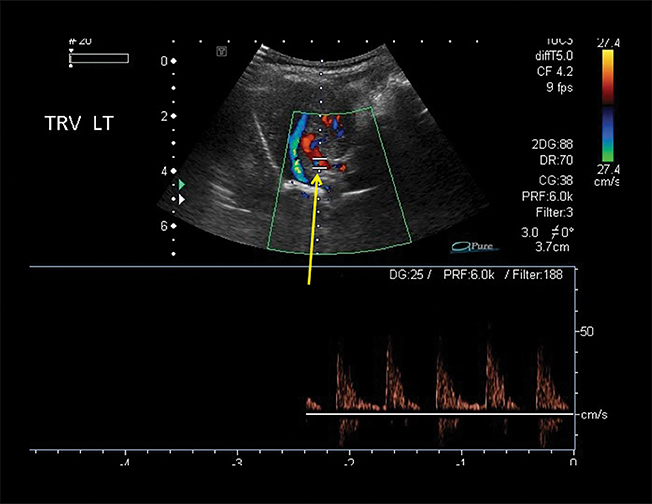
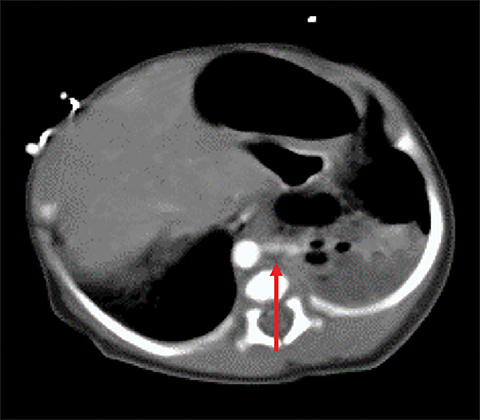
Owing to the frequent systemic arterial supply of these lesions, multidetector CT of the chest extending caudally to the upper abdomen to include the celiac axis and suprarenal fossae is the modality of choice for investigation of these lesions.11 Patients may present with a persistent focal opacity or focal cystic change, which enhances after contrast administration and has a systemic arterial supply (Figure 4). The pathologic manifestations of the cystic portion in many of these lesions are now recognized to be similar to the pathology of type 2 CPAMs. This recognition has led to the more recently used diagnostic term “hybrid” lesion (Figure 5).
Intra-abdominal sequestrations have uncommonly been found just cephalad to the kidney. MRI use in evaluating sequestrations has been described anecdotally; however, many children would require sedation and intravenous gadolinium.
Risks Associated with CLMs
Risk of infection and malignancy, and the potential for CLM regressionin asymptomatic patients, play a key role in influencing management. Advances in our understanding of CLMs with respect to these factors have led to the recent, although still controversial, trend to question the previously held notion that most, if not all, cystic lung malformations require excision.13-16 Indeed, the quality of the literature addressing these factors is still suboptimal.17, 18
Data on the risk of infection of CLMs is variable, ranging widely from 3% to more than 80% through multiple studies.14,16,19 For instance, in a 2018 study,20 13 of 39, or 33%, of initially asymptomatic patients with CLMs who were being followed conservatively, subsequently required resection after becoming symptomatic. In contrast, a 2010 study describes only 5% of CPAMs resected for recurrent infection.21
A rare risk of malignancy is increasingly associated with some CPAMs, with case reports prior to 1980 suggesting a connection between congenital cystic malformations and rare pulmonary tumors. This literature was anecdotal and did not receive the type of methodological scrutiny now applied to most publications. However, the subsequent classification of a previously unappreciated cystic pulmonary tumor, a pleuropulmonary blastoma (PPB), shed some light on the original literature and suggested some of these anecdotal reports may have been describing a PPB.22
Even Stocker, in his more recent classification schema,10 admitted that the newer “Type 4“ CPAM was almost identical in pathological appearance to a PPB. A genetic DICER1 mutation is now known to result in an autosomal dominant tumor predisposition syndrome, seen in approximately 70% of all PPB cases. The mutation also increases the risk for developing cystic nephromas of the kidneys, Sertoli-Leydig cell tumors of the ovaries, thyroid tumors, and rhabdomyosarcomas.
Unfortunately, differentiating a PPB from a CPAM with CT alone is difficult. Both lesions contain cystic change within the lung, and both can have more solid appearing areas on CT (Figure 6). A recent review of more than 100 cases of each23 could only produce findings more suggestive of one or the other. The presence of a pneumothorax, more complex cystic and solid areas, and the presence of a DICER1 mutation were more predictive of a PPB, while prenatal diagnosis before 30 weeks,24-26 the presence of a systemic feeding vessel, and an area of associated overinflated lung parenchyma were more predictive of a congenital malformation.
Anecdotal reports of a rare but definite association between Type 1 CPAMs (containing cysts >2 cm) and bronchioalveolar carcinoma (BAC) in young adults have also emerged.27-29 A review of the pathology of the cysts in these CPAMs has revealed a distinct premalignant potential of the cellular lining of these larger cysts.30-31 Mucinous cells present in many cases of CPAM type 1 may serve as precursors to BAC, and apparently type 1 CPAMs express MUC5AC, similar to that seen in adults with BAC.32 While these case reports are scattered and rare, there is sufficient evidence to support the assertion that type 1 CPAMs likely have a premalignant potential, although the risk remains .
CLM Management
Prenatally, CLMs should be followed with ultrasound, with MRI reserved for high-risk lesions when intervention is being considered or the diagnosis is unclear.18 Postnatally, radiography may be of benefit in symptomatic patients, but its value in the asymptomatic child is questionable. Contrast-enhanced CT, recommended to characterize the lesion, provides critical information to influence the decision of whether to pursue surgical intervention.11-12
The size of CPAMs and sequestrations has the potential to regress. Postnatal follow-up CTs have been shown to identify lesions that are occult on prenatal US and follow-up radiographs. Additionally, longitudinal studies have demonstrated that CLOE grows more slowly than the normal lung, taking up a progressively smaller portion of the affected hemithorax over time.33-34
Debate persists regarding the optimal pre- and postnatal management strategy of cystic CLMs, despite recent growing understanding of the etiology and pathology of these entities. Attempting to guide practice, the American Pediatric Surgery Association (APSA) has published evidence-based recommendations on CPAM treatment, but the group cautions no existing literature meets Level I or Level 2 evidence criteria.18 Prenatal intervention with hydrops induced by the presence of a cystic malformation is critical, as there is a high fetal mortality rate for the hydropic fetus.35 Maternal betamethasone administration36 and/or surgical in utero cyst drainage at an experienced high-risk prenatal center have resulted in fetal survival rates higher than 70%.37-38
Resection of symptomatic CLMs is widely accepted; it is practiced at our institution following postnatal contrast-enhanced CT. Resection of symptomatic CPAMs in the first year of life is still advocated, with a surgical complication rate ranging from 5% to 23%, depending on the study.17,39 APSA recommends resection before children are 6 months of age for ease of operative intervention, adequate recovery, and time for compensatory lung growth.17
Management of the asymptomatic infant with a cystic CLM remains controversial. While precedent exists for conservative CLM management, some centers continue to resect all CLMs regardless of size or complexity. If asymptomatic CLMs are to be observed, APSA recommends obtaining at least one postnatal CT examination. We typically perform this CT at 4-6 months of age and take the opportunity to attempt to establish a diagnosis and comment on regional lung changes, cyst size and complexity, mass effect, vascular supply and drainage, and any additional congenital anomalies.
Type 1 CPAMs are electively resected due to the suspected, though , risk of BAC. Many children with CLMs demonstrating features more predictive of CPAMs than PPB, such as those diagnosed prenatally, negative for DICER1 mutation, and with cysts less than 2 cm in size, are managed conservatively. Lesions that would necessitate bilateral thoracotomy or pneumonectomy have also been observed.16, 19, 20, 22
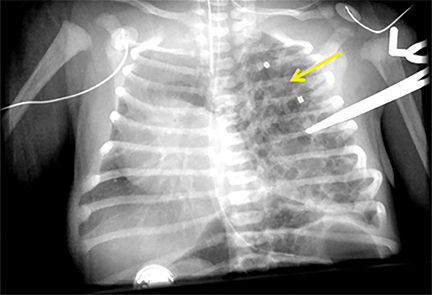
CLOE is managed according to symptomatology. Resection is only considered in symptomatic children (Figure 7). Most extralobar sequestrations are resected electively.
Conclusions
CLMs comprise a spectrum of pulmonary malformations with a proposed unifying mechanism of induction involving in utero bronchial obstruction. All children with suspected CLM should undergo contrast-enhanced CT within the first few months of life to better characterize the lesion and define its location, extent, and vascularity. As the potential exists for mixed pathology in each lesion, assignment of a pathologic diagnosis is less important than accurate description of the characteristics and anatomy of lesions and surrounding structures. Symptomatic lesions warrant elective surgical resection, unless the symptomatology warrants immediate resection. The ultimate decision to proceed with surgical resection may depend on factors beyond radiological appearance. Prenatal sonographic findings, symptom complex, DICER1-mutation testing, risk of superinfection, and malignant potential are all issues that must be addressed by the clinical team and the child’s parents/guardians.
References
- Kunisaki SM, Fauza DO, Nemes LP, et al. Bronchial atresia: the hidden pathology within a spectrum of prenatally diagnosed lung masses. J Pediatr Surg. 2006;41(1):61-65.
- Davenport M, Warne SA, Cacciaguerra S, et al. Current outcome of antenally diagnosed cystic lung disease. J Pediatr Surg. 2004;39(4):549-556.
- Riedlinger WF, Vargas SO, Jennings RW, et al. Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation, and lobar emphysema. Pediatr Dev Pathol. 2006;9(5):361-373.
- Gornall AS, Budd JL, Draper ES, et al. Congenital cystic adenomatoid malformation: accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn. 2003;23(12):997-1002.
- McCullagh M, MacConnachie I, Garvie D, et al. Accuracy of prenatal diagnosis of congenital cystic adenomatoid malformation. Arch Dis Child. 1994;;71(2):F111-113.
- Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12(1):17-37.
- Thacker PG, Rao AG, Hill JG, et al. Congenital lung anomalies in children and adults: current concepts and imaging findings. Radiol Clin North Am. 2014;52(1):155-181.
- Hislop A, Reid L. New pathological findings in emphysema of childhood. 1. Polyalveolar lobe with emphysema. Thorax. 1970;25(6):682-690.
- Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8(2):155-171.
- Stocker JT. Congenital and developmental diseases. In : Dail DH, Hammer SP, eds. Pulmonary Pathology 3rd ed. New York: Springer; 2008:154-180.
- Lee EY, Dorkin H, Vargas SO. Congenital pulmonary malformations in pediatric patients: review and update on etiology, classification, and imaging findings. Radiol Clin North Am. 2011;49(5):921-948.
- Epelman M, Kreiger PA, Servaes S, et al. Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR. 2010;31(2):141-157.
- Keidar S, Ben-Sira L, Weinberg M, et al. The postnatal management of congenital cystic adenomatoid malformation. Isr Med Assoc J. 2001;3(4):258-261.
- Singh R, Davenport M. The argument for operative approach to asymptomatic lung lesions. Semin Pediatr Surg. 2015;24(4):187-195.
- Adzick NS, Flake AW, Crombleholme TM. Management of congenital lung lesions. Semin Pediatr Surg. 2003;12(1):10-16.
- Thompson AJ, Sidebotham EL, Chetcuti PAJ, et al. Prenatally diagnosed congenital lung malformations-A long-term outcome study. Pediatr Pulmonol. 2018;53(10):1442-1446.
- Baird R, Puligandla PS2, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg. 2014;23(5):270-277.
- Downard CD, Calkins CM, Williams RF, et al. Treatment of congenital pulmonary airway malformations: a systematic review from the APSA outcomes and evidence based practice committee. Pediatr Surg Int. 2017; 33(9): 939-953.
- Aziz D, Langer JC, Tuuha SE, et al. Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation: to resect or not? J Pediatr Surg. 2004;39(3):329-334.
- Criss CN, Musili N, Matusko N, et al. Asymptomatic congenital lung malformations: Is nonoperative management a viable alternative? J Pediatr Surg. 2018;53(6):1092-1097.
- Nasr A, Himidan S, Pastor AC, et al. Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg. 2010 Jun;45(6):1086-1089.
- Vargas SO, Korpershoek E, Kozakewich HP, et al. Cytogenetic and p53 profiles in congenital cystic adenomatoid malformation: insights into its relationship with pleuropulmonary blastoma. Pediatr Dev Pathol. 2006;9(3):190-195.
- Feinberg A, Hall NJ, Williams GM, et al. Can congenital pulmonary airway malformation be distinguished from Type I pleuropulmonary blastoma based on clinical and radiological features? J Pediatr Surg. 2016;51(1):33-37.
- Waelti SL, Garel L, Soglio DD, et al. Neonatal congenital lung tumors - the importance of mid-second-trimester ultrasound as a diagnostic clue. Pediatr Radiol. 2017;47(13):1766-1775.
- Miniati DN, Chintagumpala M, Langston C, et al. Prenatal presentation and outcome of children with pleuropulmonary blastoma. J Pediatr Surg. 2006;41(1):66-71.
- Mechoulan A, Leclair MD, Yvinec M, et al . Pleuropulmonary blastoma: a case of early neonatal diagnosis through antenatal scan screening. Gynecol Obstet Fertil. 2007;35(5):437-441.
- Priest JR, Williams GM, Hill DA, et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44(1):14-30.
- Ishida M, Igarashi T, Teramoto K, et al. Mucinous bronchioloalveolar carcinoma with K-ras mutation arising in type 1 congenital cystic adenomatoid malformation: a case report with review of the literature. Int J Clin Exp Pathol. 2013;6(11):2597-2602.
- Messinger YH, Stewart DR, Priest JR, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer. 2015;121(2):276-285.
- Granata C, Gambini C, Balducci T, et al. Bronchioloalveolar carcinoma arising in congenital cystic adenomatoid malformation in a child: a case report and review on malignancies originating in congenital cystic adenomatoid malformation. Pediatr Pulmonol. 1998;25(1):62-66.
- MacSweeney F, Papagiannopoulos K, Goldstraw P, et al. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol. 2003;27(8):1139-1146.
- Lantuejoul S, Nicholson AG, Sartori G, et al. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioloalveolar carcinoma precursors. Am J Surg Pathol. 2007;31(6):961-969.
- Mei-Zahav M, Konen O, Manson D, et al. Is congenital lobar emphysema a surgical disease? J Pediatr Surg. 2006;41(6):1058-1061.
- Kovacevic A, Schmidt KG, Nicolai T, et al. Two further cases supporting nonsurgical management in congenital lobar emphysema. Klin Padiatr. 2009;221(4):232-236.
- Azizkhan RG, Crombleholme TM. Congenital cystic lung disease: contemporary antenatal and postnatal management. Pediatr Surg Int. 2008;24(6):643-657.
- Albanese CT, Sydorak RM, Tsao K, et al. Thoracoscopic lobectomy for prenatally diagnosed lung lesions. J Pediatr Surg. 2003;38(4):553-555.
- Knox EM, Kilby MD, Martin WL, et al. In-utero pulmonary drainage in the management of primary hydrothorax and congenital cystic lung lesion: a systematic review. Ultrasound Obstet Gynecol. 2006;28(5):726-734.
- Adzick NS, Harrison MR, Crombleholme TM, et al. Fetal lung lesions: management and outcome. Am J Obstet Gynecol. 1998;179(4):884-889.
- Hall NJ, Chiu PP, Langer JC. Morbidity after elective resection of prenatally diagnosed asymptomatic congenital pulmonary airway malformations. Pediatr Pulmonol. 2016;51(5):525-53.
Citation
M A, D M. Work-up and Imaging of Congenital Lung Malformations . Appl Radiol. 2020;(4):30-35.
June 30, 2020